
基于原生质体的谷子CRISPR/Cas9 基因编辑系统优化
2022-04-18 04:49刘光宇徐晓静夏科科孙海汐陶月如
河南农业科学 2022年1期
刘光宇,徐晓静,夏科科,孙海汐,陶月如,崔 震,顾 颖
(深圳华大生命科学研究院,广东 深圳 518083)
谷子(Setaria italica)作为1 年生的二倍体禾本科植物,主要在亚洲、非洲北部、南美洲和北美洲的干旱和半干旱地区种植,是优质的粮饲兼用作物[1-2]。由于谷子基因组较小、生育期短、自花授粉、种质资源丰富、与其他主粮作物和生物质能源草本植物关系密切,有巨大的潜力成为模式作物以更好地了解C4 植物的光合作用[3-4]。虽然在过去的20 a谷子的遗传转化效率较低(5.5%~9.0%),但最近高效的谷子遗传转化方法已经被报道,转化效率高达19.2%,这使得利用基因编辑工具进行谷子功能基因研究和遗传改良成为可能[5-8]。
CRISPR/Cas9 (Clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9)系统是进行基因编辑的重要工具之一。该系统由gRNA(Guide RNA)引导Cas9 在靶位点通过识别前间隔序列邻近基序(PAM)产生双链DNA 断裂,随后通过非同源末端连接(NHEJ)或同源定向修复(HDR)机制引入基因突变[9]。NHEJ 主要通过产生短小的核苷酸插入或缺失造成基因突变,而HDR介导的基因组编辑可在目标DNA 序列中精确引入所需的基因片段[10]。
CRISPR/Cas9 系统已被广泛有效地应用于包括植物在内的多种生物的靶向基因突变。将CRISPR/Cas9 系统应用于一个新的植物物种时,首先需要针对系统中的表达元件,如启动子、Cas9和gRNA 等进行优化。内源性的组成型启动子和密码子优化可以提高Cas9 在宿主内的表达水平[11-16]。gRNA 表达元件呈现多元化,比如单一gRNA、双gRNA 串联和基于tRNA 的工具包等。增加gRNA 数量可以使gRNA表达元件释放更多成熟的gRNA,增加Cas9的靶位点,进而提高基因编辑效率或进行多位点的同时编辑[17-20]。对Cas9 和gRNA 表达元件同时进行优化组合可进一步提高基因编辑效率。这些高效的基因编辑工具被用于植物育种以提高新品种的培育效率[10,21]。
植物原生质体可用于快速评估基因编辑工具的效率,进而对基因编辑工具进行优化[22-24]。植物原生质体易于获得,并且基因编辑工具可通过聚乙二醇(PEG)快速导入原生质体中,试验在7 d内可以完成。基因编辑工具通过植物组织培养的遗传转化周期至少在3个月以上,获得再生植株后还需要进行大量的筛选工作。因此,基于原生质体的方法优化CRIPSR/Cas9系统比基于组织培养的方法更高效。另外,核糖核蛋白(RNP)复合物也可以通过PEG 介导传递到植物原生质体中,在不引入外源DNA 的情况下诱导靶点突变[25-27]。这种突变区别于转基因作物的突变机制,有助于提高公众对基因编辑作物的接受度。
为了检测转化原生质体后CRISPR/Cas9系统诱导的突变频率,需要提取原生质体DNA 并对靶向序列进行PCR 扩增,随后可以用2 种简单的方法来评估扩增产物:DNA 酶分析法和大规模平行测序法。DNA 酶分析法依赖酶消化产物的凝胶图像密度来粗略估计诱变效率,包括限制性片段长度多态性(RFLP)、切割扩增多态性序列分析和T7 核酸内切酶Ⅰ(T7EⅠ)分析[28]。但由于限制性内切酶切割不完全或PCR 扩增错误等原因,很难在靶点检测到低效率的突变,并且容易出现假阳性结果,而大规模平行测序法可以通过计算突变序列的百分比来避免这些问题[29-30]。
目前,在谷子中学者们对一些简单的CRISPR/Cas9 系统已经进行了测试[22-23]。LIN 等[22]设计了一种单一RNA的基因编辑工具,将其转入谷子原生质体中,通过DNA酶分析法测得其对八氢番茄红素脱氢酶基因(PDS)靶点的编辑效率为10.2%。CHENG等[23]设计了双gRNA 系统,通过农杆菌介导法将其转入谷子愈伤组织中,在65 个再生植株中检测到26个编辑事件。但是,高效的基因组编辑工具仍需要开发和优化。因此,针对谷子内源化Cas9表达元件,设计了多种gRNA 表达元件与其组合构建了多种CRISPR/Cas9 基因编辑系统,将它们转入谷子原生质体中,通过大规模平行测序技术分析SiPDS基因的突变频率来评估这些基因编辑系统的编辑效率,从而快速筛选出基于tRNA 的高效基因编辑工具,并验证RNP 复合物在谷子中进行基因编辑的有效性。这种基于原生质体的筛选系统可用于评估和优化谷子基因组编辑工具,有助于加速谷子的基因功能研究和遗传改良进程。
1 材料和方法
1.1 试验材料及其培养
供试谷子品种为豫谷1 号,将其成熟种子置于30 ℃烘箱中48 h 打破休眠,随后浸泡于5% H2O2溶液中16 h 以提高种子萌发率,然后用75%乙醇消毒1 min,2.5%次氯酸钠表面消毒10 min。消毒完成后,挑选饱满健康的种子平铺于培养皿中的滤纸上,加入适量的蒸馏水,置于28 ℃培养箱中,光照条件下培养2 d,再转入黑暗条件下培养5 d,随后收获黄化苗用于谷子原生质体的制备[31-32]。
1.2 载体构建
从Phytozome 数据库获取谷子(Setaria italicv2.2)参考基因组序列,通过BLAST 获得谷子PDS基因序列,并进一步通过Sanger 测序确认该基因在豫谷1号中的序列信息。利用CRISPR-GE(http://skl.scau.edu.cn/)设计6 个gRNA 用以靶向SiPDS基因保守结构域,序列和位置信息见图1。根据谷子密码子偏好优化Cas9序列并在序列首尾加入2 个核定位信号(NLS)。所有核苷酸序列通过BGI-Write 平台合成。
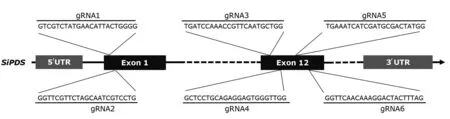
图1 设计的gRNA序列和靶向SiPDS的位置Fig.1 Designed gRNAs sequences and targeting sites in SiPDS
首先,以pCAMBIA1300 载体为骨架整合Super启动子(SP)或谷子内源的泛素启动子(Ubi)驱动的绿色荧光蛋白基因(GFP),构建报告质粒SP-GFP和Ubi-GFP以指示原生质体瞬时转化效率。然后,同样以pCAMBIA1300 载体为骨架整合SP 或Ubi 启动子驱动的Cas9 表达元件,构建中间载体SP-Cas9和Ubi-Cas9以进一步构建多种基因编辑系统。合成的单一gRNA、双gRNA 和基于tRNA 结构并包含4个gRNA 的表达元件由水稻(Oryza sativa)U6 启动子驱动,并通过多克隆位点区域的限制性内切酶BsaⅠ的2 个位点将其分别导入SP-Cas9或Ubi-Cas9中间载体,构建SP-gRNA1、Ubi-gRNA1、UbigRNA3、Ubi-dgRNAE1、Ubi-dgRNAE12、Ubi-tRNA质粒(图2)。SP-gRNA1 和Ubi-gRNA1 用以筛选在谷子中更适合驱动Cas9的启动子。Ubi-gRNA1、Ubi-gRNA3、Ubi-dgRNAE1、Ubi-dgRNAE12 和UbitRNA 用以优化gRNA 表达元件,从而筛选高效的CRIPSR/Cas9基因编辑系统。

图2 构建的CRISPR/Cas9基因编辑系统Fig.2 The constructed CRISPR/Cas9 gene editing systems
1.3 RNP复合物制备和体外切割活性分析
用MEGAshortscript™T7 转录试剂盒(Invitrogen,AM1354)进行gRNA 体外转录。参考LIANG 等[33]的方法在Cas9 反应缓冲液中将1 μg gRNA3、gRNA4和gRNA5 体外转录产物分别与1 μg Cas9(NEB,M0386S)混合并在室温下静置10 min 制备RNP 复合物。根据SiPDS基因靶位点附近的序列设计1 对引物,引物序列为5′-CCTGCAAGGTACCAACTTGA-3′和5′-GGTGCTTTTATGTGCAACCAGG-3′,通过PCR扩增获得667 bp 的扩增产物。将此扩增产物纯化后,添加制备好的RNP 复合物,37 ℃下共培养2 h。取20 μL反应液在1.5%的琼脂糖凝胶中于120 V电压下电泳20 min,然后通过观察凝胶图像检测Cas9的切割活性,以筛选高效的RNP复合物。
1.4 原生质体的制备和瞬时转化
原生质体分离和转化采用PEG 介导的改良方法进行[32]。用刀片将黄化苗的幼茎切成0.5~1.0 mm 的小段,随后放入装有15 g/L 纤维素酶‘Onozuka’R-10(Yakult Pharmaceutical)、7.5 g/L 离析酶R-10(Yakult Pharmaceutical)、10 mmol/L 吗啉乙磺酸(MES,pH 值5.7,Sigma,M3671)、10 mmol/L CaCl2(Sigma,C5670)、0.4 mol/L D-甘露醇(Sigma,M1902)和0.1% 牛血清白蛋白(BSA,Sigma,V900933)溶液的三角瓶中。三角瓶包裹锡纸后放入摇床中,60 r/min 培养约5 h 进行酶解反应。反应完成后组织悬浮液通过孔径40 μm 的过滤器去除酶解溶液和组织碎片,用W5 缓冲液[154 mmol/L NaCl(Sigma,S5886)、5 mmol/L KCl(Sigma,P5405)、125 mmol/L CaCl2和2 mmol/L MES(pH 值5.7)]清洗2 次。将清洗后的混合溶液以100×g转速缓慢离心5 min,收集原生质体,随后用W5 缓冲液再洗涤原生质体2次,并在MMG 溶液(0.4 mmol/L D-甘露醇、15 mmol/L MgCl2和4.0 mmol/L MES,pH 值5.7)中重悬原生质体,显微镜下用血细胞仪计数,然后将细胞浓度调整至2×106个/mL。
质粒(10 μg)和体外切割活性较高的RNP 复合物(20 μg Cas9 和20 μg gRNA)分别与细胞浓度为2×106个/mL 的原生质体在新鲜配制的400 g/L PEG溶液中暗置20 min。除去上清液后重悬于1 mL W5缓冲液中,室温暗置培养48 h。
1.5 大规模平行测序
提取室温暗置培养48 h 的原生质体DNA,并设计引物(表1)扩增含有SiPDS基因靶位点的DNA 序列。使用MGIEasy AmpSeq 建库试剂盒构建测序文库。用BGISEQ-500测序仪对文库进行大规模平行测序。
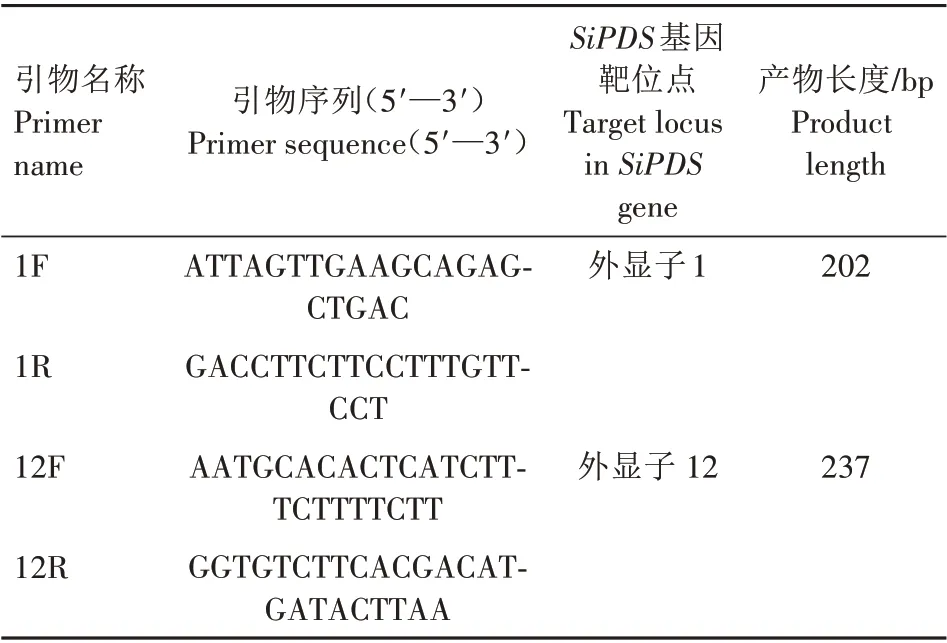
表1 测序文库构建所用引物信息Tab.1 Primers used for construction of sequencing library
测序read 中的突变有4 种类型,包括缺失、插入、替换和混合(替换加缺失、替换加插入等复杂类型)。每种类型的总突变效率通过突变的read 总数除以总read 数来估计。为了尽量减少扩增子测序错误引起的问题,测序数据中只发生一次的突变read 不计算在内[31]。每个CRISPR/Cas9 基因编辑系统的诱变效率由每个gRNA 序列区域内所有突变类型的总突变效率决定。
2 结果与分析
2.1 高效谷子原生质体转化系统的建立
为了建立高效的谷子原生质体转化系统,利用PEG 介导法将SP-GFP和Ubi-GFP质粒转入来源于7 d 黄化苗幼茎的原生质体中。质粒中的GFP基因表达的产物受到紫外或蓝光激发时会发射绿色荧光(图3a)。通过显微观察和统计发现,SP-GFP和Ubi-GFP质粒的转化效率为50.44%和57.36%(图3b),这与之前报道的高效谷子原生质体转化系统的转化效率相近[22,34]。
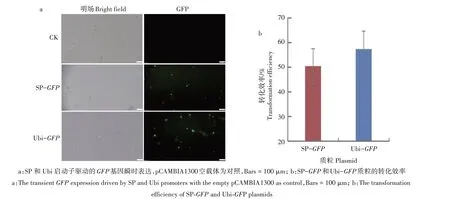
图3 SP和Ubi驱动GFP基因在谷子原生质体中的瞬时表达Fig.3 The transient expression of GFP driven by SP and Ubi promoters in S.italica protoplasts
2.2 谷子原生质体中多种CRISPR/Cas9基因编辑系统的优化
为了筛选更适合驱动Cas9基因表达的启动子,构建了SP-gRNA1 和Ubi-gRNA1 质粒并检测它们在谷子原生质体中对SiPDS基因的编辑效率。结果表明,转SP-gRNA1 质粒的谷子原生质体的基因突变频率为0.5%,而转Ubi-gRNA1 质粒的谷子原生质体的基因突变频率为5.5%(图4),说明在谷子原生质体中内源性Ubi 启动子比SP 启动子更适合驱动Cas9基因的表达。因此,在后续试验中选择Ubi启动子来驱动Cas9基因的表达。

图4 CRISPR/Cas9 基因编辑系统对SiPDS基因的突变频率Fig.4 Mutation efficiency of SiPDS gene induced by CRISPR/Cas9 gene editing systems
为了研究多元化gRNA 表达元件对提高基因突变效率的作用,设计了双gRNA系统,并构建了UbidgRNAE1 和Ubi-dgRNAE12 质粒。与单一gRNA 系统Ubi-gRNA1 相比,Ubi-dgRNAE1 对SiPDS基因第一外显子靶点的基因突变频率增加了1.66 倍,为14.56%(图4)。靶向SiPDS基因第12 外显子的双gRNA 系统Ubi-dgRNAE12 比单一gRNA 系统UbigRNA3 的基因突变频率提高了1.11 倍,为15.71%(图4)。这些结果表明,额外添加1 个gRNA 可以提高CRISPR/Cas9 基因编辑系统对SiPDS基因的突变效率。
为了进一步提高靶向基因的突变效率,利用tRNA 结构优化gRNA 表达元件并构建了包含4 个gRNA 的Ubi-tRNA 质粒。结果显示,转Ubi-tRNA质粒的谷子原生质体的SiPDS基因突变频率提升至51.24%,相较于转Ubi-gRNA3 和Ubi-dgRNAE12 质粒的谷子原生质体分别增加了5.87 倍和2.26 倍(图4)。综上,利用tRNA 系统组装多个gRNA 可以进一步提高CRISPR/Cas9 基因编辑系统对SiPDS基因的突变效率。
2.3 谷子原生质体中SiPDS基因多个靶位点同时编辑的实现
为了进一步调查多元化gRNA 基因编辑系统诱导SiPDS基因的多位点突变效率,本研究统计了Ubi-dgRNAE1、Ubi-dgRNAE12、Ubi-tRNA 系统在单一靶位点和多个靶位点同时发生编辑事件的频率。结果(表2)表明,Ubi-dgRNAE1 在单一靶位点的突变频率为14.42%,而同时编辑2 个靶位点的突变频率仅为0.14%。在转Ubi-dgRNAE12 谷子原生质体中,也发现同时编辑SiPDS基因2 个靶位点的突变频率极低。上述结果表明,利用双gRNA 系统同时编辑SiPDS基因2 个靶位点的效率极低。转Ubi-tRNA 谷子原生质体中SiPDS基因单一靶位点的突变频率为48.00%,同时编辑2、3 个靶位点的突变频率分别为2.99%、0.24%,分别占总突变频率的5.83%、0.47%。这些结果证实了tRNA 结构可以有效提高基因编辑系统同时靶向SiPDS基因多个位点的突变效率,而双gRNA 串联结构对提高多位点编辑效率的作用不明显。
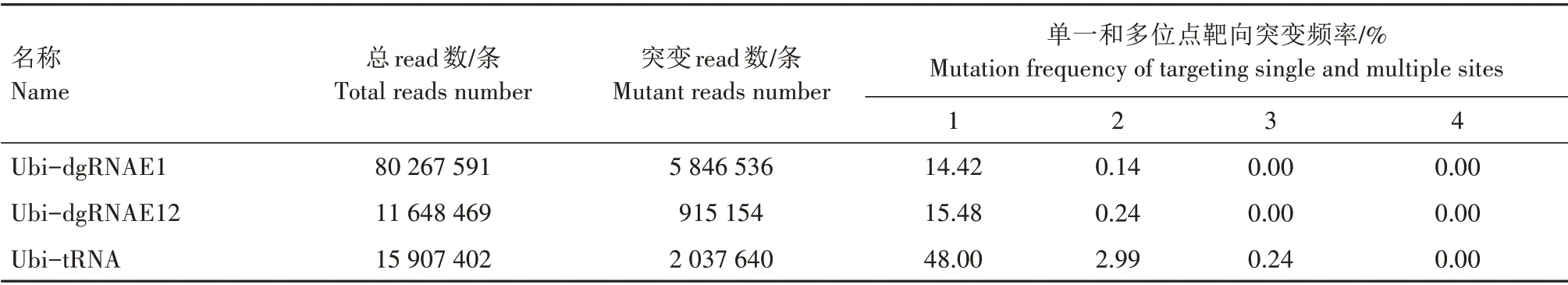
表2 谷子原生质体中CRISPR/Cas9基因编辑系统同时靶向SiPDS基因多个位点的突变频率Tab.2 Mutation frequency of targeting multiple sites by CRISPR/Cas9 gene editing systems in S.italica protoplasts
2.4 RNP复合物在谷子原生质体中编辑SiPDS基因的有效性分析
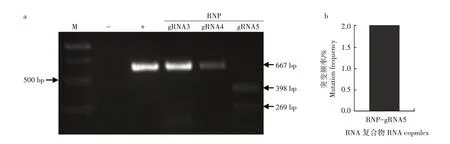
为了筛选高效的RNP 复合物,选取gRNA3、gRNA4、gRNA5 进行体外转录,其产物分别与Cas9蛋白混合制备RNP 复合物。用RNP-gRNA3、RNPgRNA4 和RNP-gRNA5 复合物对SiPDS基因靶点扩增的DNA序列进行体外切割活性检测,凝胶图像显示RNP-gRNA5 复合物体外切割活性高于RNPgRNA3和RNP-gRNA4复合物(图5a)。根据此结果将RNP-gRNA5 复合物转入谷子原生质体后进行大规模平行测序以评估其引起SiPDS基因突变的效率。结果表明,RNP-gRNA5复合物引起SiPDS基因的突变频率为2.0%(图5b)。
为了进一步研究RNP-gRNA5 复合物诱导的SiPDS基因靶点突变类型,对突变谱进行了分析,包括1、2、3 及以上碱基对(bp)缺失、插入和其他(替换、替换加缺失、替换加插入等)突变类型。统计结果(图5c)显示,缺失突变类型在总突变类型中的占比高达98.33%。其中,1、2、3 bp 缺失突变类型分别占35.41%、35.30%、24.76%,3 bp以上缺失突变类型占比下降至2.86%。插入和其他突变类型在总突变类型中的占比不超过2%。上述结果表明,RNPgRNA5 复合物诱导的SiPDS基因突变类型主要为小片段(1~3 bp)的缺失。
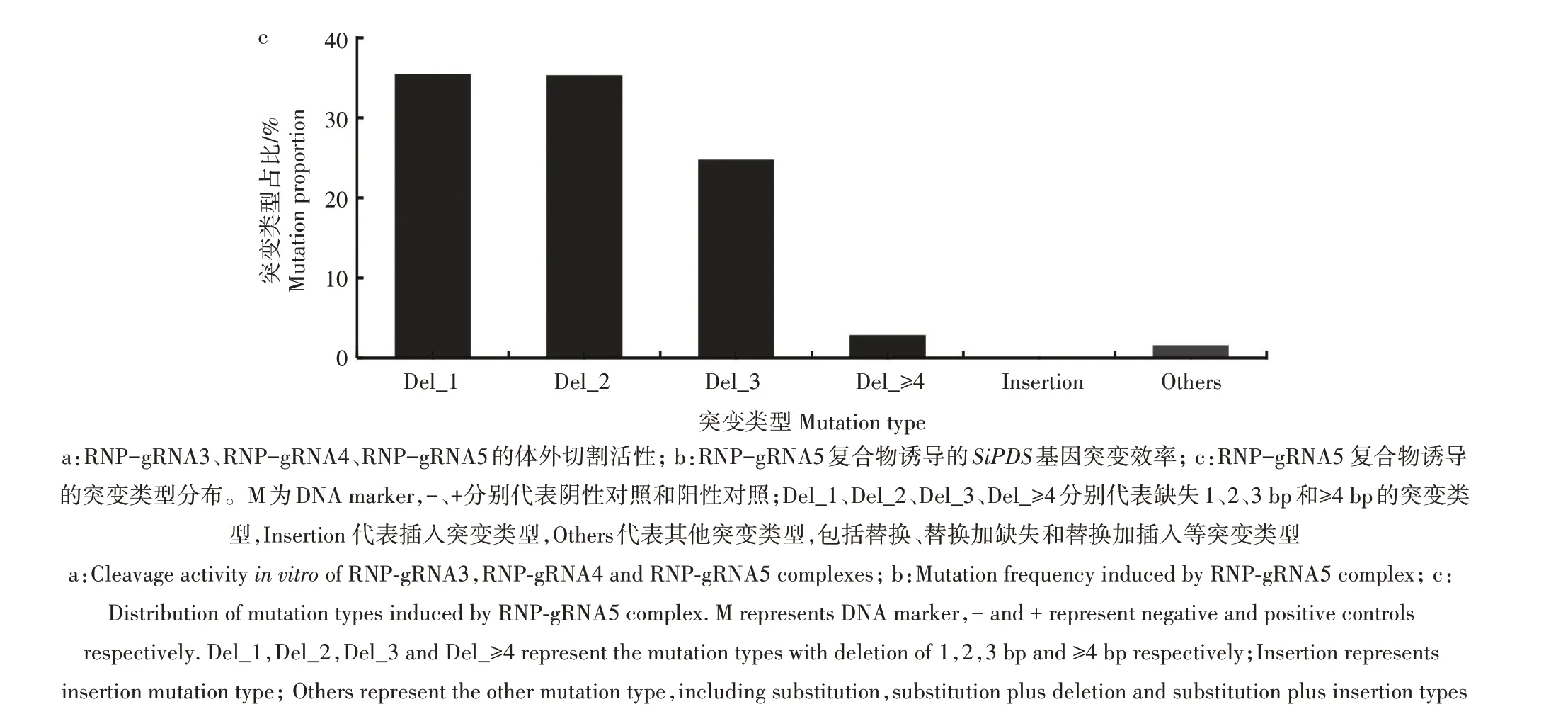
图5 RNP复合物在谷子原生质体中编辑SiPDS基因的有效性分析Fig.5 Efficiency of RNP complexes editing SiPDS gene in S.italica protoplasts
3 结论与讨论
谷子稳定的遗传转化可以实现编辑位点在后代植株中的稳定遗传,但需要组织培养、载体传递和植株再生等较烦琐的过程,对于基因编辑工具的筛选和优化既费时又费力。相对而言,谷子原生质体更容易获得,转化容易,试验周期较短,已用于突变检测[22]、再生[35]和蛋白质互作[36]等研究中。与前人[22,34-36]研究结果相比,本研究采用生长7 d 的黄化苗制备谷子原生质体,从时间上更早,并且转化效率可以达到同样的高效水平。此外,大规模平行测序可以在短时间内以较低的人工成本实现高通量突变检测和分析,并且可以提供比传统DNA酶分析法更准确的统计数据。因此,基于谷子原生质体的大规模平行测序系统可以作为一种简便有效的方法对基因编辑工具进行快速筛选和优化,在稳定遗传转化前即可筛选到高效的基因编辑工具。
CRISPR/Cas9 系统在谷子中的应用相对比较简单,目前只有包含单一或2 个gRNA 的基因编辑工具被报道,并且其基因突变效率不高[22-23]。CRISPR/Cas9 系统中更多的组件需要测试和优化以开发更高效的基因编辑工具。优化Cas9基因的密码子以适应寄主植物已成为共识。许多研究用内源性Cas9 启动子提高CRISPR/Cas9 系统的基因突变效率[13-16]。本研究利用开发的原生质体系统证明谷子内源性Ubi 启动子可以提高CRISPR/Cas9 基因编辑系统对SiPDS基因的突变效率,也可以进一步应用本研究开发的这套系统快速筛选更高效的启动子及其他元件。
多元化的gRNA 表达元件可以释放出更多成熟的gRNA,以靶向多个位点从而提高编辑效率。2个gRNA串联的表达元件可以提高Cas9在胡萝卜细胞中的靶向概率[18],相似的表达元件被用来靶向猕猴桃PDS基因的2 个不同外显子以确保基因敲除[19]。本研究结果表明,与单一gRNA 导向的基因编辑系统相比,2 个gRNA 串联的基因编辑系统UbidgRNAE1 和Ubi-dgRNAE12 的总突变频率提高,但是同时编辑2 个靶位点的的突变频率还是极低,说明在CRISPR/Cas9 系统中串联2 个gRNA 的表达元件对同时编辑2 个靶位点效率的提升并不明显,在最近报道的研究中也发现了类似的结果[23]。进一步利用tRNA 结构优化CRISPR/Cas9 系统,构建的Ubi-tRNA 质粒不仅具有较高的总诱变频率,而且增强了多重基因组编辑能力。这些结果表明,基于tRNA 的CRISPR/Cas 基因编辑系统可以高效地引起目标基因突变,提高诱变效率。未来将其应用于谷子的稳定遗传转化可能产生更多的突变植株,甚至可以进行多基因编辑以达到更高的育种目标。
通过RNP 复合物进行基因组编辑提供了一种在植物中无外源DNA 引入和低脱靶编辑的方法[10]。在作物中应用RNP 复合物引起的编辑效率并不高,介于2.4%~9.7%,但在莴苣中却异常地高达46%[25-27]。本研究结果表明,RNP 复合物RNPgRNA5 在谷子原生质体中的诱变效率为2.0%。这种相对较低的诱变效率可能是由于RNP-gRNA5 的转化效率较低或者是选取的gRNA 活性不高。不过这些问题可以通过使用本研究开发的原生质体系统进一步筛选优化更多的RNP 复合物来解决。尽管RNP 复合物引起的基因突变效率不高,但由于该方法简便、快速、可操作性强,对于大多数研究来说仍然是可以接受的[26]。此外,RNP 复合物可在不引入外源DNA 的情况下对目标基因进行编辑。RNP复合物有巨大的潜力成为重要的基因编辑工具。
猜你喜欢
中国生物化学与分子生物学报(2022年6期)2022-09-06
中国典型病例大全(2022年11期)2022-05-13
中国典型病例大全(2022年7期)2022-04-22
中草药(2022年5期)2022-03-03
中国生物化学与分子生物学报(2022年1期)2022-02-26
当代陕西(2021年21期)2022-01-19
科学导报(2021年29期)2021-06-03
科海故事博览·下旬刊(2019年6期)2019-04-16
延河(2018年6期)2018-06-11
湖南师范大学学报·自然科学版(2014年5期)2014-11-14
