
2017-2019年贵州省BV系流感病毒的分子特征分析
2022-04-25 12:57万永虎任丽娟郑勤妮蒋维佳雷明玉李世军牟鸿江
中国人兽共患病学报 2022年4期
万永虎,任丽娟,郑勤妮,庄 丽,蒋维佳,雷明玉,李世军,牟鸿江,郭 华
流感是呼吸道传播的高度传染性疾病,每年导致全球近50万人死亡[1],严重威胁着人类健康。B型流感病毒每年都能造成季节性流行、暴发或大范围疫情,并能引发多种并发症和死亡病例,还能产生很大的经济与社会影响,导致的疾病负担甚至超过A型[2-4],故B型流感病毒也不可低估。国家流感中心曾对我国69个代表株进行检测,发现了12个重配株[5]。贵州省每年监测点采集流感样本14 000份左右,近6年阳性率平均为13.46%,B型占26.49%,所占比例较高,且B型为优势流行株时,此期间流感流行强度往往较高,暴发疫情也多由B型流感导致,据研究统计显示,2013-2017年贵州省共报告疫情149起,B型46起,占30.87%,且主要的系别就是BV系流感病毒[6];贵州省统计数据显示,2017-2018年流感主要以季节性H3和新甲型H1N1为主要流行型别,BV系暴发疫情较少,但2019年报告了81起暴发疫情,BV系流感病毒导致了39起,占48.15%,重症病例8例,其流行形势十分严峻。流感易变异,尤其是表面糖蛋白基因HA和NA基因的变异直接影响到病毒的致病力、毒力和耐药性,人群对变异株普遍易感,暴发疫情和重症死亡病例的发生往往是变异毒株在人群中的表现,而及时地、更好地了解流感病毒的分子特征对流感的防控至关重要[7]。然而,贵州省BV系流感病毒的变异情况、与疫苗株的匹配性以及是否耐药目前尚不清楚。
本文拟对贵州省近年BV系流感病毒暴发疫情和重症病例毒株HA和NA基因的遗传特征与分子变异情况、国内外代表株与疫苗株间的异同和对神经氨酸酶类抑制剂药物的耐药性进行分析,以期了解贵州省BV系流感病毒的分子流行特征与变异情况,及时发现变异株、重配株,以及探索暴发疫情和重症病例毒株的特异性分子靶标;了解其与疫苗株的匹配性;明确其对药物的敏感性,及时发现耐药株。为贵州省流感防控策略的制定、疫苗的选择、疫苗免疫效果的评价和新疫苗的研制及临床用药提供科学依据。
1 材料与方法
1.1 材料来源 选取贵州省2017-2019年BV系流感暴发疫情毒株16株,含重症病例毒株3株,另外普通流行株8株,经RT-PCR扩增出9株HA和NA基因序列,连同国家疾控中心测序提供的15株毒株序列共计24株,以及WHO推荐的疫苗株2株,共计26株进行HA和NA基因的分子特征分析。毒株暴发疫情毒株编号和名称依次为:4:B/Guizhou/Qixinguan/1729/2017;5:B/Guizhou/Qixinguan/1781/2017; 13:B/GZ/Bijiang/1516/2019;14:B/GZ/bijiang/1267/2019;15:B/Guizhou/Zhenyuan/33/2019;16:B/Guizhou/Chishui/53/2019;17:B/Guizhou/Honghuagang/1392/2019;18:B/Guizhou/Honghuagang/1965/2019;19:B/Guizhou/Pingba/35/2019;20:B/Guizhou/Huichuan/1150/2019;21:B/GZ/zhongshan/1345/2019;22:B/GZ/zhongshan/1363/2019;23:B/GZ/zhongshan/1381/2019;重症病例毒株:24:B/GZ/Zunyi/YQ12/2019;25:B/GZ/Zunyi/YQ13/2019;26:B/GZ/Zunyi/YQ32/2019;普通流行毒株:3:B/Guizhou/Qixinguan/326/2017; 6:B/Guizhou/Qingzhen/140/2018;7:B/Guizhou/Douyun/11123/2018;8:B/Guizhou/Honghuagang/1214/2018;9:B/Guizhou/Qixinguan/1443/2019;10:B/GZ/xinyi/1269/2019;11:B/Guizhou/Honghuagang/1438/2019;12:B/Guizhou/Kaili/1346/2019;疫苗株为:1:B/Brisbane/60/2008(2017年WHO推荐的BV系疫苗株)和2:B/Colorado/06/2017 (2018-2019年WHO推荐的BV系疫苗株),疫苗株和其余毒株参考序列均下载至美国国家生物技术信息中心和全球流感基因共享数据库。
1.2 HA和NA基因的扩增 应用磁珠法Ex-DNA/RNA病毒核酸提取试剂盒(西安天隆科技有限公司),按照说明书操作进行样本基因组的提取,然后使用特异性引物BHA-M13F8:TGTAAAACGACGGCCAGTGCAGAGCATTTTCTAA, BHA-M13R1885:CAGGAAACAGCTATGACC AGTAGTAACAAGAGCAT;BNA-M13F1:TGTAAAACGACGGCCAGTAGCAGAAGCAGAGC-
ATCTTC,BNA-M13R1556:CAGGAAACAGC-TATGACCAGTAGTAACAAGAGCAT,并用Prime Script One Step RT-PCR Kit Ver.2试剂盒(宝日医生物技术(北京)有限公司)扩增HA和NA基因核苷酸序列,扩增产物大小HA和NA基因分别为1 749 bp和1 479 bp,产物经1.0%的琼脂糖凝胶电泳鉴定,大小符合后,委托生工(上海)生物工程有限公司进行HA和NA基因双端测序,测序结果已提交至NCBI,基因序列号为OL545372-OL545389。
1.3 HA和NA基因分子特征解析 应用生物信息学软件包Lasergene7.1中的EditSeq、SeqMan软件对HA和NA基因测序结果进行编辑和处理,使用MEGA7.0软件进行序列比对,MegAlign软件进行HA和NA基因核苷酸同源性图表的制作,再采用MEGA7.0软件进行HA和NA基因氨基酸位点的解析,并用Neighbor-Joining法Bootstrap重复值设置为1 000构建分子遗传进化树。
2 结 果
2.1 HA和NA基因遗传进化分析 将贵州省2017-2019年BV系流感暴发疫情、重症病例、普通流行株和疫苗株以及同源性比对最高的毒株一起构建HA和NA基因分子遗传进化树,结果显示:贵州省所有毒株HA和NA基因分子遗传进化树拓扑结构类似,整体上毒株处于3个分支。所有暴发疫情、重症病例毒株HA和NA基因与2018-2019年推荐的疫苗株B/Colorado/06/2017遗传距离相对2017年之前推荐的疫苗株B/Brisbane/60/2008遗传距离更近,但2019年的毒株与2018-2019年推荐的疫苗株已经存在一定的遗传距离。另外,2017年七星关区暴发疫情毒株HA和NA基因与七星关区普通流行毒株遗传距离较近,聚集为一簇;2018年的毒株分散存在没有聚集成簇,存在一定的遗传差距;2019年的毒株HA和NA基因均处于两个分支,分别为分支1与分支2且相对较远,重症病例毒株均处于分支1,且分支1中平坝县、钟山区、惠水县、遵义(重症病例毒株)、红花岗区和赤水县暴发疫情毒株与贵州毕节七星关区、兴义和遵义红花岗区以及四川、美国密西西比地区普通流行毒株HA和NA基因遗传距离最短,亲缘关系最近并占主要部分;分支2中碧江区和镇远县暴发疫情毒株与黔东南州凯里普通流行毒株遗传距离最短,亲缘关系最近。提示2019年贵州省BV系毒株HA和NA基因存在较大变异,存在两个分支的毒株流行且流行株主要与分支1中的毒株为主。详见图1、图2。
2.2 HA和NA基因同源性分析 将贵州省2017-2019年BV系流感病毒暴发疫情、重症病例毒株同疫苗株和普通流行毒株一起进行核苷酸同源性比对,结果显示:贵州省所有毒株同疫苗株B/Brisbane/60/2008 HA和NA基因的同源性分别为97.4%~98.9%和98.2%~98.6%,同疫苗株B/Colorado/06/2017的同源性分别为98.6%~99.3%和98.9%~99.4%,提示与新推荐的疫苗株同源性更高;另外2017年BV系流感病毒毒株间HA和NA基因同源性分别为99.9%和99.9%~100%;2018年毒株间同源性为98.4%~99.3%和98.7%~99.1%;2019年毒株间同源性分别为97.7%~100%和98.4%~99.9%,其中2019年分支2中的4株毒株之间同源性分别为99.4%~100%和99.6%~99.9%,分支1中的15株间同源性分别为99.5%~100%和99.3%~99.9%,提示2019年毒株同源性差距较大,变异较多,主要分为两群。2017年暴发疫情毒株同当年度普通流行毒株之间HA和NA基因同源性均为99.9%;2018年无BV系暴发疫情;2019年分支2中的暴发疫情毒株与当年度普通流行株之间同源性分别为99.4%和99.6%~99.7%,分支1中的暴发疫情毒株含重症病例毒株与当年度普通流行株之间同源性均为99.5%~99.9%。提示暴发疫情、重症病例毒株与普通流行株之间同源性相对较高。详见表1、表2。

△:represent outbreak strains;▲:represent severe pneumonia cases strains;●:represent vaccine strains图1 2017-2019年贵州省BV系流感病毒HA基因遗传进化树Fig.1 Phylogenetic tree of the HA genes of influenza BV lineage strains in Guizhou in 2017-2019
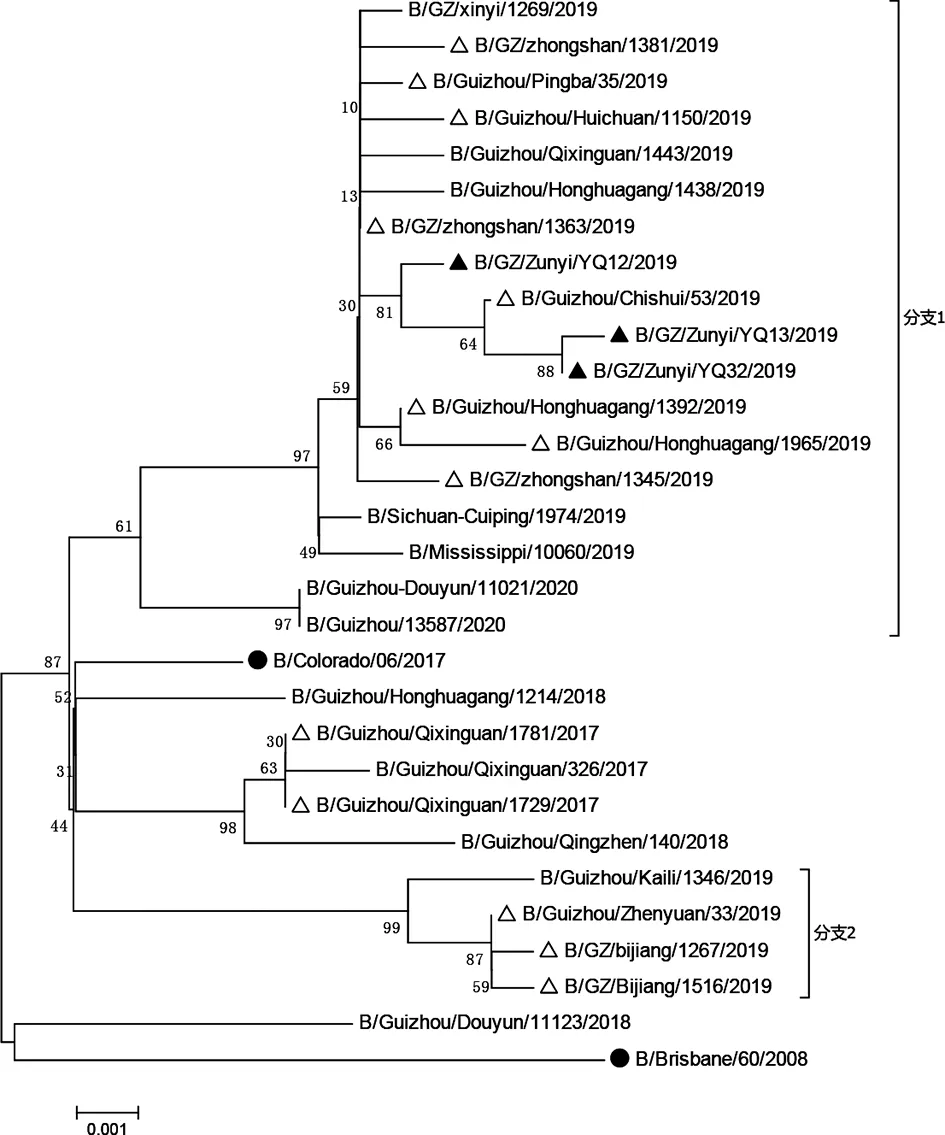
△:represent outbreak strains;▲:represent severe pneumonia cases strains;●:represent vaccine strains图2 2017-2019年贵州省BV系流感病毒NA基因遗传进化树Fig.2 Phylogenetic tree of the NA genes of influenza BV lineage strains in Guizhou in 2017-2019

表1 贵州省BV系流感病毒HA基因同源性Tab.1 Sequence identity of the HA genes of influenza BV lineage strains in Guizhou

表2 贵州省BV系流感病毒NA基因同源性Tab.2 Sequence identity of the NA genes of influenza BV lineage strains in Guizhou
2.3 HA和NA基因分子特征变异分析 乙型流感病毒HA基因含有4个主要的抗原决定簇,分别为120环(116-137位点)、150环(141-150位点)、160环(162-167位点)和190螺旋(194-202位点)。氨基酸序列变异分析显示:与2017年之前推荐的疫苗株B/Brisbane/60/2008相比,贵州省BV系毒株HA和NA基因分别存在20个和32个氨基酸位点的突变,其中HA基因所有毒株均存在的突变有3个,分别为I132V、N144D、S212N,N144D位点涉及到150环,NA基因所有毒株均存在的突变有6个,分别为K220N、S295R、N340D、D384G、N470S、V481I;与2018-2019年推荐的疫苗株B/Colorado/06/2017相比HA和NA基因分别存在22个和27个氨基酸位点的突变,其中HA基因所有毒株均存在的突变有4个,分别为S14N、G144D、V195I、K513R,G144D和V195I位点分别涉及到150环和190螺旋,NA基因所有毒株均存在的突变有2个,分别为Q371K、D490N。与其他毒株相比,2017-2018年毒株HA基因无特异性位点突变,NA基因2017年和2018年部分毒株存在I45T、I115V、P124T、V422I 4个特异性位点突变;2019年处于进化树分支1中的14株毒株HA基因存在G148R、K151E、T563A 3个特异性位点突变,G148R位点涉及到150环,NA基因存在A395T的特异性突变;处于进化树分支2中的4株毒株HA基因存在V394I的特异性突变,NA基因存在V71A、K343E、A395V、V401I 4个特异性位点的突变。更为重要的是,进化树分支1中的14株毒株与B/Brisbane/60/2008相比HA基因存在179-181KND 3个位点的特异性缺失突变,与B/Colorado/06/2017相比存在181D一个位点的特异性缺失突变。2017年暴发疫情毒株HA基因与普通流行毒株相比无突变,NA基因存在A91T、S266F突变;2018年无暴发疫情;2019年暴发疫情、重症病例毒株与普通毒株之间HA基因存在个别突变,无特异性突变,仅进化树分支2中NA基因存在T46I突变。耐药位点分析结果显示,所有毒株均存在G402S的突变,其余耐药位点均较稳定,未发生突变。详见表3、表4。

表3 贵州省BV系流感病毒HA基因氨基酸位点变化Tab.3 HA gene mutations affecting amino acids of influenza BV lineage strains in Guizhou
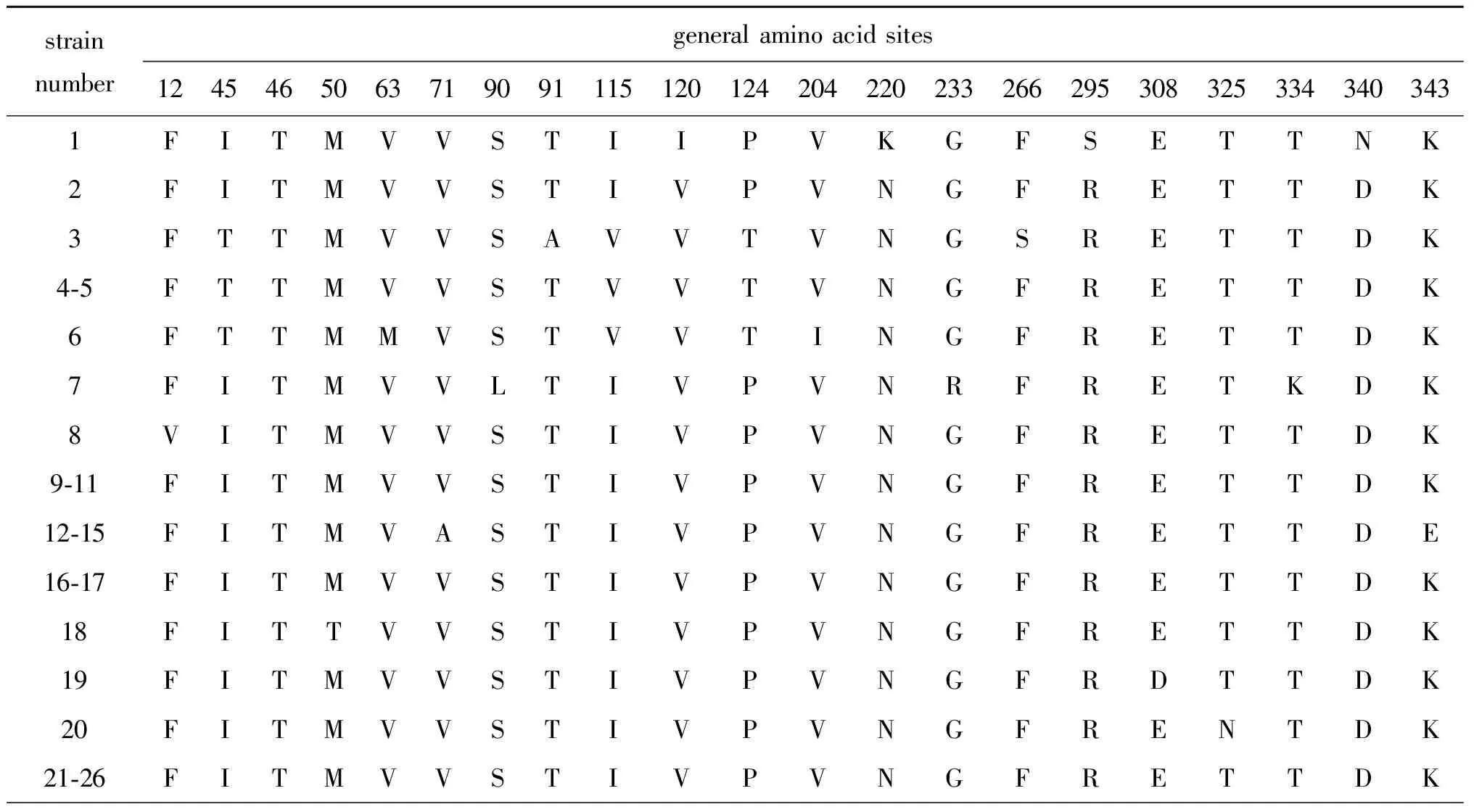
表4 贵州省BV系流感病毒NA基因氨基酸位点变化Tab.4 NA gene mutations affecting amino acids of influenza BV lineage strains in Guizhou
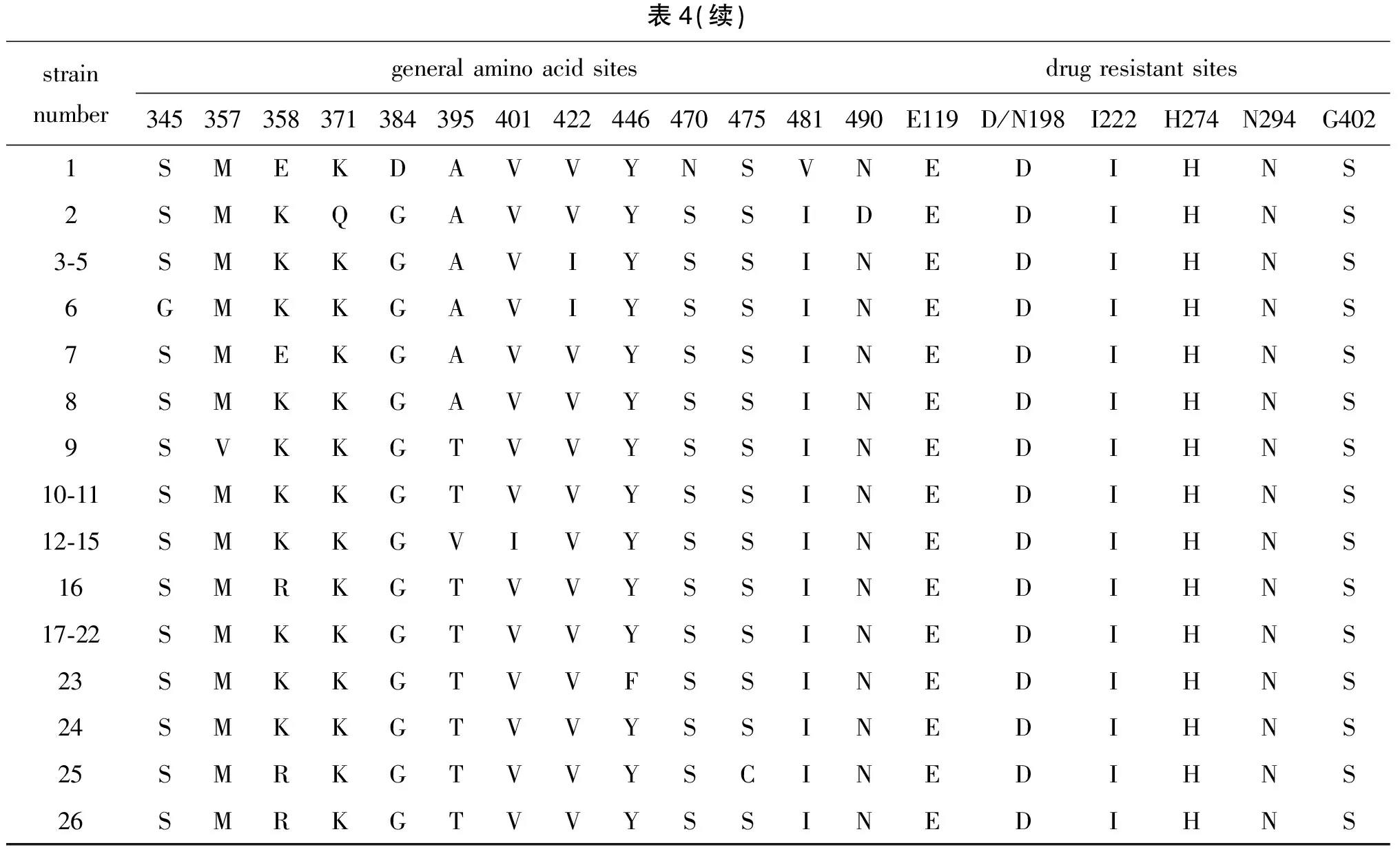
表4(续)strain numbergeneral amino acid sitesdrug resistant sites345357358371384395401422446470475481490E119D/N198I222H274N294G4021SMEKDAVVYNSVNEDIHNS2SMKQGAVVYSSIDEDIHNS3-5SMKKGAVIYSSINEDIHNS6GMKKGAVIYSSINEDIHNS7SMEKGAVVYSSINEDIHNS8SMKKGAVVYSSINEDIHNS9SVKKGTVVYSSINEDIHNS10-11SMKKGTVVYSSINEDIHNS12-15SMKKGVIVYSSINEDIHNS16SMRKGTVVYSSINEDIHNS17-22SMKKGTVVYSSINEDIHNS23SMKKGTVVFSSINEDIHNS24SMKKGTVVYSSINEDIHNS25SMRKGTVVYSCINEDIHNS26SMRKGTVVYSSINEDIHNS
3 讨 论
乙型流感病毒可通过抗原漂移和抗原转变两种机制来逃避宿主的免疫应答和促进自身的进化,抗原漂移是其主要的方式,即流感病毒蛋白尤其是HA和NA蛋白发生微小的突变,如点突变的积累或者序列的插入、缺失从而导致流感病毒抗原性的改变,这种突变常常引起中小型流感暴发的流行[8],如1985年加拿大、1988年美国和1993年中国乙型流感的暴发流行均是由病毒抗原漂移导致,2006年我国河北省流行的BV系流感病毒也是由HA1区域发生抗原漂移变异产生的一个新变种所致[9]。另外,基因的重配也是流感病毒进化的重要方式,即不同来源的流感病毒基因片段组合在一起形成新的病毒,如2006年我国浙江省乙型流感的大范围流行[10]。
暴发疫情的发生常常是毒株变异的表现。贵州省此阶段研究的所有毒株HA和NA基因分子遗传进化树拓扑结构类似,未发现HA和NA基因重配现象。流感流行株与疫苗株间HA基因是否匹配是影响疫苗保护效果的重要因素,据国家流感中心统计资料显示,如果流行株与疫苗株不匹配,人群保护效果将从70%~90%降低到20%~30%。贵州省此阶段研究的所有暴发疫情、重症病例毒株HA和NA基因与2018-2019年推荐的疫苗株B/Colorado/06/2017遗传距离更近,同源性更高,提示新推荐的疫苗株保护效果可能更好,但2019年的毒株与此疫苗株之间已经存在一定的遗传距离和同源性差异,提示我们随着毒株的不断变异需进一步及时更新更加匹配的疫苗株。2017年暴发疫情毒株、2019年分支1和分支2中暴发疫情毒株处于3个分支,分别与相应的普通流行毒株HA和NA基因遗传距离最短,亲缘关系最近,同源性均较高,提示贵州省BV系暴发疫情毒株之间存在一定的差距,但暴发疫情毒株与普通流行毒株之间亲缘关系和同源性较高,整体上贵州省2019年BV系毒株存在两个分支群系毒株的流行,且暴发疫情、重症病例毒株主要以遗传进化树分支1中的毒株流行为主,分支1中的毒株可能成为今后BV系毒株中的优势流行毒株,应进一步加强该分支毒株的生物学特性监测和研究。
贵州省BV系暴发疫情、重症病例毒株HA和NA基因分子特征变异显示,与2017年之前推荐的疫苗株B/Brisbane/60/2008相比,贵州省BV系毒株HA基因所有毒株均存在I132V、N144D、S212N的突变,N144D涉及到抗原决定簇的150环,NA基因均存在K220N、S295R、N340D、D384G、N470S、V481I的突变;与2018-2019年疫苗株B/Colorado/06/2017相比HA基因均存在S14N、G144D、V195I、K513R的突变,G144D和V195I位点分别涉及到150环和190螺旋,NA基因均存在Q371K、D490N的突变。提示贵州省BV系毒株HA和NA基因与新推荐的疫苗株相比氨基酸突变数和有效累积突变数都少,但涉及到两个抗原决定簇区域的突变,可能为新的流行株。李多等[11]在研究2017年云南省BV系毒株与2018-2019年疫苗株B/Colorado/06/2017相比时,HA基因仅均存在S14N和V195I两个位点的突变,而我省新增加了G144D和K513R,也提示我省BV系毒株的有效累积突变更多或毒株变异更强。另外,与其他毒株相比,我省2017-2018年毒株仅NA基因2017年和2018年部分毒株存在I45T、I115V、P124T、V422I 4个特异性位点突变;2019年进化树分支1中的毒株HA基因存在G148R、K151E和T563A 特异性位点突变,且G148R位点涉及到抗原决定簇150环,NA基因存在A395T的特异性突变;进化树分支2中的毒株HA基因存在V394I的特异性突变,NA基因存在V71A、K343E、A395V和V401I 特异性位点的突变。尤其是进化树分支1中的毒株HA基因与疫苗株B/Brisbane/60/2008相比存在179-181KND 3个位点的特异性缺失突变,与B/Colorado/06/2017相比存在181D一个位点的特异性缺失突变。氨基酸的插入和缺失会使基因编码的产物长度发生变化,从而对蛋白的功能造成影响,陈继明等[12]对1972-2000年我国乙型流感病毒HA1区的基因特征分析发现,HA1区曾发生过多次核苷酸的插入和缺失,但仅发生在氨基酸162-166位,故我省应加强乙型流感病毒分子特征的实时监测。
贵州省暴发疫情毒株与普通流行株之间比对发现,2017年暴发疫情毒株与普通流行毒株相比仅NA基因存在A91T、S266F突变;2018年无暴发疫情;2019年暴发疫情、重症病例毒株与普遍毒株之间HA基因存在个别突变,无特异性突变,仅进化树分支2中毒株NA基因存在T46I突变;推测贵州省2019年BV系流感暴发疫情的发生可能与179-181KND 3个位点的特异性缺失突变和HA基因G148R、K151E、T563A以及NA基因A395T的特异性突变有关,但需进一步研究确定。
有研究表明[13-14],与乙型流感病毒耐药相关的突变位点有E119、R156、W178、S179、D/N198、I222、E227、H274、E277、N294和G402。这些位点的突变可能导致病毒对药物敏感性的下降,贵州省耐药位点分析结果显示,所有毒株均存在G402S的突变,其余耐药位点均较稳定,未发生突变,需进一步明确其对神经氨酸酶抑制剂类药物的有效性。
总之,加强乙型流感病毒尤其是暴发疫情、重症死亡病例毒株致病力的特异性分子靶标研究,有利于高效准确地评估病毒的致病力大小和预测其流行潜能,为流感防控策略的制定、疫苗的选择和研制以及临床用药提供科学依据。
利益冲突:无
引用本文格式:万永虎,任丽娟,郑勤妮,等. 2017-2019年贵州省BV系流感病毒的分子特征分析[J]. 中国人兽共患病学报,38(4):366-373. DOI:10.3969/j.issn.1002-2694.2022.00.012
猜你喜欢
动物医学进展(2022年9期)2022-11-26
临床医学工程(2022年3期)2022-04-20
科学大观园(2022年2期)2022-01-23
文萃报·周二版(2021年47期)2021-12-14
祝您健康·文摘版(2019年2期)2019-06-11
科学24小时(2019年5期)2019-06-11
教师·中(2018年4期)2018-06-02
中国医学创新(2017年7期)2017-03-31
家庭百事通·健康一点通(2017年3期)2017-03-22
江苏农业科学(2016年8期)2017-02-15
