
RET抑制剂Pralsetinib的研究进展
2022-04-26 08:16王小涵代洪爽崔永生
辽宁大学学报(自然科学版) 2022年1期
陈 烨,王小涵,王 铭,代洪爽,崔永生
(1.辽宁大学 药学院,辽宁 沈阳 110036;2.沈阳城市学院 商学院,辽宁 沈阳 110112)
0 引言
癌症是人类主要死因之一[1-2],其特征是细胞分裂异常并扩散到身体不同部位.这种疾病是由负责维持细胞功能的基因受损引起的[3].基因受损会导致细胞功能改变,并可能使细胞恶化.在不同的癌细胞中都可以识别出转染重排(rearranged during transfection,RET)基因,如非小细胞肺癌、甲状腺乳头状癌、甲状腺髓样癌、结肠癌和其他实体肿瘤[4].RET是一种众所周知的原癌基因,RET原癌基因编码一种跨膜受体酪氨酸激酶[5-6],在神经系统和肾脏的胚胎发育中具有生理作用,这类分子还包括TRK-A、TRK-B、TRK-C、EGFR和PDGFR-(1,2).RET的激活改变与许多实体肿瘤的分子发病机制有关,包括甲状腺癌和非小细胞肺癌.RET基因的染色体重排发生在编码RET胞内激酶结构域序列与另一个蛋白的n-末端蛋白二聚体结构域序列相连时[7-8],这些重排通常允许RET在转录沉默的细胞中异常表达,并导致致癌的细胞内RET融合.在1%~2%的非小细胞肺癌(NSCLC)患者中,RET受体酪氨酸激酶的染色体重排是致癌的驱动因素.美国食品药品监督管理局(FDA)最近批准了选择性RET酪氨酸激酶抑制剂普拉替尼(Pralsetinib),用于治疗转移性RET融合阳性NSCLC[9].
1 Pralsetinib的合成
Pralsetinib的合成途径专利由原研公司所持有,并根据其专有的化合物库所设计[10].图1为原研公司在化合物专利中的合成路线.
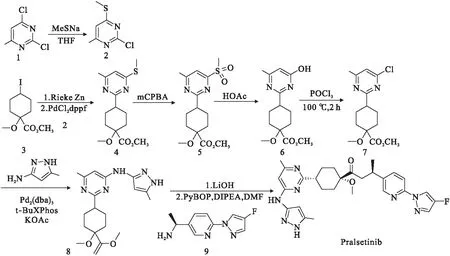
图1 普拉替尼(Pralsetinib)国外合成路线[10]
图2为国内专利报道的一条关于Pralsetinib的合成路线,该发明[11]涉及卤代、脱甲基保护和酰胺缩合3步反应,减少了反应步骤,提高了反应总收率,有效地降低了反应终产物的成本.
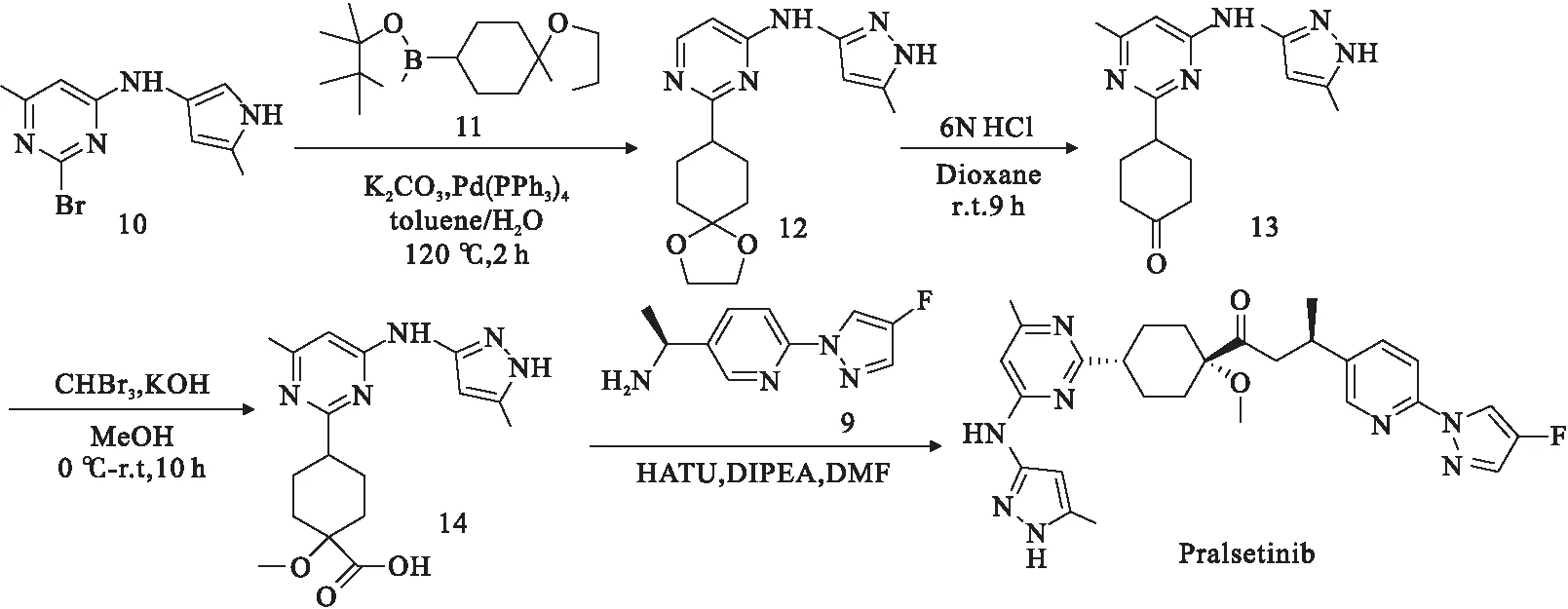
图2 普拉替尼(Pralsetinib)国内专利合成路线[11]
2 Pralsetinib的药效
Pralsetinib(以前称为BLU-667)是一种口服酪氨酸激酶抑制剂[12],对包括V804 gatekeeper突变在内的致癌RET融合和突变具有选择性和强靶向性,与其他酪氨酸激酶相比对RET表现出高选择性.在多种临床中,Pralsetinib对RET改变的肿瘤模型显示了抗肿瘤活性[13].
Pralsetinib暴露-反应关系和药效学反应的时间过程尚未完全确定.在体外酶检测中,Pralsetinib有效地抑制了野生型和激活的RET突变体(IC50 0.4 nmol/L)[14],对血管内皮生长因子受体2(VEGFR-2)有88倍的选择性.在4~15 nmol/L浓度下,该药物抑制RET驱动的肿瘤细胞系包括MZ-CRC-1(RET M918T,MTC)、TT(RET C634W,MTC)、TPC-1(PTC)和LC2/ad(NSCLC)细胞的自磷酸化和增殖[15].在体内,口服Pralsetinib与多种小鼠异种移植模型中的致癌RET激酶活性的剂量依赖性抑制相关,包括RET(C634W)突变的甲状腺髓样瘤、患者来源的KIF5B-RET NSCLC、患者来源的CCDC6-RET和CCDC6-RET(V804M)结直肠癌[16],后者包含一个gatekeeper突变,可对多种激酶抑制剂产生耐药性,包括普纳替尼(Ponatinib)、卡博替尼(Cabozantinib)和凡德他尼(Vandetanib).与Cabozantinib相比,Pralsetinib可抑制肿瘤生长[17].

表1 ARROW疗效结果[21]
Pralsetinib是一种有效的、选择性的RET抑制剂,可靶向RET的改变,包括融合和突变.Pralsetinib具有特异性[18],对RET融合突变具有高选择性,同时对其他激酶具有较低的亲和力.Pralsetinib具有广泛和持久的抗肿瘤活性,可用于多种晚期实体肿瘤的治疗[19].Pralsetinib耐受性良好,在所有接受治疗的人群中其安全性是一致的[20].
在一项多中心、非随机、开放标签、多队列临床试验(ARROW,NCT03037385)中研究人员评估了Pralsetinib对RET融合阳性转移性NSCLC患者的疗效,Pralsetinib对患者表现出较好的抗肿瘤活性[21].该研究在单独的队列中纳入了接受铂类化疗进展的转移性RET融合阳性NSCLC患者和Treatment-Naïve转移性NSCLC患者,并根据实体肿瘤的疗效评价标准(RECISTv1.1),通过盲法独立中心审查(BICR)进行评估,主要疗效结果指标包括客观缓解率(ORR)和缓解持续时间(DOR).表1总结了Pralsetinib对87例既往接受过铂化疗的RET融合阳性NSCLC患者疗效结果.对于39例接受抗PD-1或抗PD-L1治疗的患者,患者无论是顺序还是同时接受铂类化疗,探索性亚组分析的ORR为59%(95% CI:42,74),中位DOR未达到(95% CI:11.3,NE).另外,有8例患者在BICR评估的基线时发现有可测量的CNS转移(这些患者在研究开始前2个月内没有接受过脑部放疗),这8例患者中有4例的颅内病变有反应,而且其中的2例CNS为完全反应.BICR评估还表明75%应答者的DOR≥6个月.表2总结了Pralsetinib对纳入ARROW的27例未接受治疗的RET融合阳性NSCLC的治疗结果,结果显示患者的ORR达到70%,58%应答者的DOR为6个月.

表2 ARROW 的测试结果(Treatment-Naïve metastatic RET-positive NSCLC)[21]
最近,RET激酶溶剂前沿顶部的L730V/I突变被鉴定为对Pralsetinib有强烈的耐药性,但对塞尔帕替尼(Selpercatinib)没有耐药性.Selpercatinib能有效抑制这些突变体和KIF5B-RET(L730V/I)致癌基因驱动的肿瘤[22].然而最近的一些研究已经确定了获得性Selpercatinib耐药的RET突变位于溶剂前(G810C/S/R),铰链(Y806C/N),以及RET atp结合位点的β2链(V738A),其中G810C/S/R突变表现出最强的耐药性[23-24],并且在肿瘤对Selpercatinib产生耐药性的患者中更常见.迄今发现的耐Selpercatinib的RET突变对Pralsetinib交叉耐药[25].
ARROW研究的结果显示,无论既往治疗如何,Pralsetinib对肿瘤都具有良好的耐受性[26],对RET融合阳性的NSCLC患者同样具有临床活性,包括颅内反应,既往接受过铂类化疗患者的应答率为61%,未接受标准治疗患者的应答率为70%.
3 药物代谢动力学
在禁食条件下,每日一次400 mg的Pralsetinib产生稳定的最大观察血药浓度(cmax)和24 h药时曲线下面积(AUC)分别为2 830 ng/mL和43 900 ng/mL.3~5 d后血药浓度达到稳定状态,每日重复口服1次后平均积累比小于2.Pralsetinib单次给药60~600 mg后,达到峰值浓度的中位时间(tmax)为2~4 h[27].
与禁食状态相比,单次给予200 mg剂量的Pralsetinib和高脂肪膳食,cmax和AUC值分别增加104%和122%,tmax从进食4 h延迟到进食8.5 h.体外血浆蛋白结合率为97.1%,与浓度无关.
经Pralsetinib单次和多次治疗后血浆消除半衰期分别为14.7 h和22.2 h.稳态表观清除率为9.1 L/h.Pralsetinib代谢途径多样,是小鼠CYP3A4的适度底物,而不是人CYP3A4的适度底物,因此CYP3A4可能并不会明显地限制Pralsetinib的口服可用性或组织分布.而外排转运体ABCB1和ABCG2能有效运输Pralsetinib,影响Pralsetinib在体内的生物利用度和分布[28].ABCB1和ABCG2在血脑屏障(BBB)的脑毛细血管内皮细胞中高表达,其外源性毒性化合物[29]可保护中枢神经系统(CNS).相反,由于这些转运体而使大脑接触到的抗癌药物很有限,可能会降低其治疗效果,特别是对脑转移瘤的治疗效果[30-33].由于ABCB1和ABCG2活性显著限制Pralsetinib对野生型大脑的渗透,使用双重抑制剂Elacridar可在一定程度上增加Pralsetinib的脑蓄积,提高Pralsetinib脑治疗效果.
在群体药代动力学研究中发现(N=405),Pralsetinib的药代动力学特性不受年龄、性别、体重、轻度、中度肾损害和轻度肝损害的临床影响[34].目前未见严重肾损害或中度或重度肝损害患者的报道.在体外研究中,Pralsetinib在临床相关浓度下可抑制CYP2C8、CYP2C9和CYP3A4/5[35],但不抑制CYP1A2、CYP2B6、CYP2C19和CYP2D6[36].Pralsetinib是P-糖蛋白和乳腺癌抵抗蛋白的底物[37],而不是胆汁盐流出泵(BSEP)、有机阳离子转运体(OCT)1、OCT2、有机阴离子转运多肽(OATP)1B1、OATP1B3、多药毒素挤压(MATE)1、MATE2-k、有机阴离子转运体(OAT)1或OAT3的底物.Pralsetinib是P-gp、BCRP、OATP1B1、OATP1B3、OAT1、MATE1、MATE2-K和BSEP的抑制剂,而不是OCT1、OCT2和OAT1A3的临床相关浓度的抑制剂.
每日1次共同口服200 mg剂量的Pralsetinib、P-gp和强CYP3A抑制剂伊曲康唑时会使Pralsetinib的cmax和AUC∞分别在体内增加84%和251%.所以,应尽量避免Pralsetinib与强CYP3A抑制剂或P-gp与强CYP3A抑制剂的联合使用[38].如果不能避免P-gp和强CYP3A抑制剂的共同使用,则应减少Pralsetinib的剂量[39].
同时,每日1次共同口服400 mg剂量的Pralsetinib与600 mg强CYP3A诱导性药物利福平时会使Pralsetinib的cmax和AUC∞分别降低30%和68%.因此,也应避免Pralsetinib与强CYP3A诱导性药物共同给药,如果仍旧无法避免联合用药的情况,则应适当增加Pralsetinib的剂量.
4 毒副作用
Pralsetinib是口服的胶囊制剂,在使用该药品时,建议每日1次空腹口服400 mg,直到疾病好转或出现毒性[37].一旦出现不良反应,应立即减少服用剂量.在I/II期ARROW研究的Pralsetinib总体安全人群中(N=438),最常见的不良反应是谷草转氨酶(AST)升高(34%),贫血(24%),谷丙转氨酶(ALT)升高(23%),便秘(23%)和高血压(22%).治疗中出现相关不良反应的情况占总治疗量的4%[38].
在ARROW实验(N=220)中,每日1次服用Pralsetinib 400 mg治疗RET融合阳性NSCLC的不良反应(≥15%)包括疲劳、发热、水肿、便秘、腹泻、口干、肌肉骨骼疼痛、高血压、咳嗽和肺炎.与基线相比恶化的实验异常包括AST水平升高,ALT水平升高,肌酐水平升高,碱性磷酸酶水平升高,钙水平下降,钠水平下降,磷酸盐水平下降,血红蛋白水平下降,淋巴细胞水平下降,中性粒细胞水平下降和血小板水平下降.严重不良反应发生在45%RET融合阳性的ARROW接受者中,最常见的是败血症、肺炎、尿路感染和发热[26].致命的不良反应发生在5%的患者中(发生在1例以上的患者包括肺炎(N=3)和脓毒症(N=2)).15%的患者出现不良反应(包括肺炎(1.8%)、肺炎(1.8%)和脓毒症(1%)),需要永久停用.60%接受Pralsetinib治疗的ARROW患者由于肺炎、中性粒细胞减少、贫血、高血压、发热、AST水平升高、血肌酸磷酸激酶水平升高、疲劳、白细胞减少、血小板减少、呕吐、ALT水平升高、脓毒症和呼吸困难等不良反应而中断.
5 展望
基于现有临床实验,RET融合已经被认为是NSCLC治疗的一个确定靶点.在2020年9月上市的RET抑制剂Pralsetinib因其对机体耐受性好、特异性高且具有持久的抗肿瘤活性,是目前已发现药物中治疗转移性RET融合阳性NSCLC最有效的药物.然而,口服使用Pralsetinib也出现了肌肉骨骼疼痛、高血压、贫血等毒副作用,且Pralsetinib暴露-反应关系和药效学反应的时间过程也未完全确定,还有待进一步研究.
猜你喜欢
肝博士(2022年3期)2022-06-30
现代临床医学(2022年3期)2022-06-06
健康体检与管理(2022年4期)2022-05-13
中国典型病例大全(2022年12期)2022-05-13
中国药学药品知识仓库(2022年5期)2022-04-11
皮肤病与性病(2021年3期)2021-07-30
现代临床医学(2021年2期)2021-03-29
科学24小时(2018年1期)2018-01-10
现代养生·下半月(2016年6期)2016-10-21
