
Treacher Collins综合征患儿临床特征及致病基因分析
2022-05-30 07:01冯英秋周妮娜刘清莲金占国
中国听力语言康复科学杂志 2022年3期
冯英秋 周妮娜 刘清莲 金占国
Thompson[1]首先报道了Treacher collins综合征(treacher collins syndrom,TCS)的症状和体征,Treacher Collin[2]首次将其定义为颧骨和下眼睑缺损综合征,该病新生儿的发生率约为1/5000[3],为一种常染色体显性遗传病[4]。由于基因突变导致的颅面骨发育不全形成特征性鱼样面容,此外还伴有颧骨与下颌发育不良、唇裂、巨口、外耳道闭锁等常见临床体征,基因突变定位于染色体5q32-5q33.1的TCOF1基因上[5], 目前已发现250余种突变位点[6]。有文献报道POLR1D和POLR1C的基因突变也可导致TCS[7,8]。本研究为门诊1例疑似TCS散发病例进行基因突变检测明确诊断,并对该病的临床特征和发病机制进行了分析和总结。
1 资料与方法
1.1 研究对象
患儿,男性,5个月,北京籍社区卫生辖区患者,足月剖宫产,因双耳听力筛查未通过,到社区全科门诊就诊。
1.2 方法
1.2.1 听力学及体格检查 全身及耳科专科检查,纯音听阈应用B.K.2231型听力计(丹麦)进行检测,声导抗测试应用Madsen 901(丹麦)检查,耳声发射检测应用Madsen CAPELLA(丹麦)进行耳蜗外毛细胞功能检测,听性脑干诱发电位潜伏期及阈值检测应用Intelligent Hearing Systems SmartEP(美国)对听神经通路进行评估。此外,还进行了头颅和颞骨CT、四肢X线检查。
1.2.2 外周血DNA提取 抽取患儿及其父母的外周静脉血,用试剂盒上海华舜生物工程有限公司提供提取法提取基因组DNA。
1.2.3 引物设计与扩增测序 TCOF1基因定位在5q32,该基因含有27个外显子,共编码1289个氨基酸,翻译treacle蛋白。在目标基因27外显子的上、下游各50 bp内含子序列区域设计相应引物(上海英骏生物技术有限公司合成),对目的基因组DNA片段进行扩增。用聚合酶链反应(polymerase chain reaction, PCR)检测技术对TCOF1基因27个外显子进行扩增,对PCR扩增产物进行纯化,直接测序。
1.2.4 TCOF1基因PCR检测体系 见表1、图1。

图1 TCOF1基因扩增PCR检测反应方案(每循环降0.5℃)

表1 TCOF1基因扩增PCR检测方案
2 结果
2.1 体检结果
患儿精神状态和营养状态良好,全身体检见:躯干和四肢发育正常、四肢活动良好、颧骨、下颌骨发育稍差、双下眼睑向外下方低垂,双耳廓发育基本正常、外耳道通畅、鼓膜完整、标志清楚,对声音反应稍迟钝。
2.2 听力学及影像学检查结果
2.2.1 听力学检查 声导抗左耳“A”型、右耳“C”型测试曲线;脑干听觉诱发电位潜伏期:双耳Ⅲ、Ⅴ波潜伏期明显延长,左侧V波潜伏期为8.63 ms,右侧V波潜伏期为7.88 ms;脑干听觉诱发电位双耳阈值均为70 dB nHL;双耳各频率均未引出有意义的畸变产物耳声发射。
2.2.2 影像学检查结果 颞骨CT示双侧外耳道通畅,中耳、内耳未见畸形;头颅MRI示脑组织发育无异常;四肢X线均未见异常,见图2。
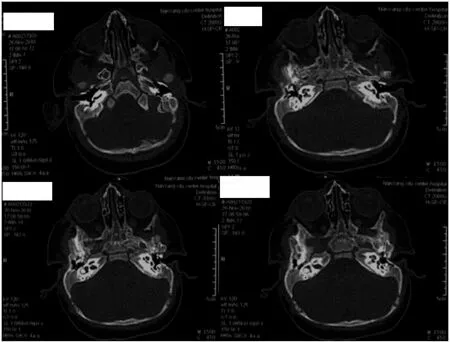
图2 患者一的颞骨CT
2.3 TCOF1基因突变筛查结果
TCOF1基因PCR后测序结果提示第2和24外显子存在杂合突变,分别为c.146C>T和c.4169C>T,第1个突变位点将编码的第49位异亮氨酸(I)转变成了苏氨酸(T)(见图3);第2个突变位点将编码的第1390位丙氨酸(A)转变成缬氨酸(V)(见图4)。患儿父母与其进行比对分析,第2个外显子未见突变,母亲的第24个外显子发现与患儿相同的突变位点(图4-b、4-c)。在NCBI网站上对TCOF1基因单核苷酸多态性(single nucleotide polymorphism,SNP)数据库中筛查发现,第24个外显子的c.4169C>T的杂合突变为SNP,而第2个外显子c.146C>T杂合突变未提示为SNP。

图3 患儿和父母TCOF1基因第2个外显子测序结果

图4 患儿和父母TCOF1基因第24个外显子测序结果
2.4 突变位点多物种保守性分析
用Clustal X软件对TCOF1基因第2外显子突变位点所处密码子编码的异亮氨酸在多物种间的保守性进行对比,发现其在不同进化程度8个物种的同源基因所编码蛋白质序列高度保守(见图5)。

图5 多物种间氨基酸序列对比
3 讨论
本研究来源于社区全科门诊就诊的一例颧骨和下颌骨发育不良、下眼睑向外下方低垂、双耳听力筛查未通过患儿。就这一少见的综合征患儿而言,首要工作是明确临床诊断,根据患儿典型的临床体征,首先考虑是与其表型相似的TCS[9]。该综合征的致病基因是TCOF1(存在78%~93%的可能)[10],基因测序发现该基因的第2个外显子出现了c.146C>T的突变位点,编码的异亮氨酸(I)变成了苏氨酸(T),进行8个物种间的保守性分析发现第49位异亮氨酸高度保守。
TCOF1基因编码核仁磷酸化蛋白Treacle[11],该蛋白在胚胎发育期间在第一、二腮弓中高表达[12]。TCOF1基因突变会致使treacle蛋白功能不全而发生TCS[13]。Treacle蛋白在颅面部神经嵴细胞发育时发挥重要作用,是胚胎发育过程中颅面部的关键蛋白,TCOF1基因突变使胚胎发育过程中神经嵴细胞迁移产生第一、二腮弓发生畸形。动物实验研究发现神经嵴细胞和神经上皮细胞的核糖体合成受阻是TCOF1基因突变导致第一二腮弓畸形的发生机制。Treacle蛋白是人类Nop65p核糖核蛋白复合物的组成成分和细胞核核糖体RNA前体形成早期的2’羟基甲基化相关联[14,15],在RNA的N-末端具有一个与核仁蛋白Lis1同源的超二级结构LisH,与微管动力学、染色体分离和细胞迁移相关[16,17],因此判断Treacle蛋白在核糖体生成过程中的某个特定阶段发挥调控功能,这种调控功能发生于细胞核和细胞质穿梭过程中,这一机制可因基因突变遭到破坏导致TCS的发生,这可能就是TCOF1基因突变的发病机理。TCOF1基因突变小鼠的神经外胚层及神经嵴细胞内成熟核糖体的量交野生型小鼠的量变少[18],以此推断,Treacle蛋白除定位细胞核的作用外,在神经上皮活性调节、核糖体发育及神经嵴细胞增殖上也发挥重要作用[18,14],TCOF1基因单倍剂量不足可诱导氧化应激反应而致DNA损伤和神经上皮细胞凋亡[19]。
TCS患者中有近50%出现传导性听力损失,主要为基因突变导致中耳腔发育不全及听小骨畸形所致[20,21],此外还存在双侧颧骨发育异常,可累及颞骨和上下颌骨,主要临床表现见表2。TCS患者中出现外耳耳廓及小耳畸形的发生率为60.0%~80.9%不等[22,23],其可分为不规则型、单侧型、顿挫型、不完全型和完全型5种类型[24]。
通过患者的临床表现、听觉功能测试和影像学检查结果对TCS的明确诊断提供参考价值。影像学检查可发现表2中的各类骨性发育异常[25,26]。利用羊膜腔穿刺抽羊水进行产前诊断可明确高度怀疑或已明确突变位点的家系怀孕成员携带致病基因情况,此外,孕检超声检查也可对胎儿发育情况进行初步判断。TCS患者约有55%出现双耳听力损失,听骨链缺如的纯音听阈测试结果多为平坦型传导性损失的听力曲线,而平坦型或斜坡型听力曲线则多为听骨链粘连或听小骨发育不全所致。

表2 TCS常见的典型临床症状
TCS治疗原则为骨骼支架重建和软组织修复。Trainor等[27]对TCS序贯治疗进行了总结报道,6岁以后可对耳廓发育异常的患儿行耳廓再造成形术,同样对大于6岁中耳发育异常患儿进行人工听骨植入,达到有效改善听力损失的目的,针对这类患儿应尽早配戴助听器并进行语言培训。下颌骨发育不良产生的小下颌畸形会产生口咽腔容积变小、舌后坠等异常,引起阻塞性睡眠呼吸暂停综合征,如存在明确的气导狭窄影响呼吸应尽早放置鼻咽通气管或进行气管切开,并在6~10岁进行颌骨成形手术,以缓解患儿阻塞性呼吸困难。Waitzman等[28]认为最好在10岁后进行颅眶颧骨重建,重建组织采用自体骨进行整复。针对外眦形态异常、耳部和鼻部急性等软组织缺损畸形,可根据异常程度择期进行整形修复,特征性的外眦角异常可上移外眦角并修复缺损眼睑,尽可能修复到可接受的程度。
本研究旨在分析1例TCS患儿致病基因的突变位点,总结该病的临床表现、发病机制及对诊疗方案,为该病开展精准诊断、临床治疗提供依据。
猜你喜欢
分子催化(2022年1期)2022-11-02
分子诊断与治疗杂志(2022年9期)2022-10-09
中国农业科学(2022年16期)2022-09-19
作物学报(2022年3期)2022-01-22
中国听力语言康复科学杂志(2021年6期)2021-12-21
电脑知识与技术(2018年19期)2018-11-01
中学生理科应试(2017年6期)2017-09-27
湖北农业科学(2014年11期)2014-09-10
