
MC2R基因变异致家族性糖皮质激素缺乏症1例
2022-06-06 03:14袁高品张晓红张丰丰陈婷丽
福建医科大学学报 2022年2期
袁高品, 张晓红, 张丰丰, 陈婷丽
家族性糖皮质激素缺乏症(familial glucocorticoid deficiency, FGD)是一种罕见的以孤立的糖皮质激素缺乏而盐皮质激素活性正常为特征的常染色体隐性遗传病。该病是由于肾上腺皮质束状带对促肾上腺皮质激素(adrenocorticotropic hormone, ACTH)不敏感而使皮质醇水平低下,ACTH明显增高,也称ACTH不敏感综合征。目前主要有3种亚型:由黑皮质素2受体(melanocortin 2 receptor, MC2R)基因致病变异引起的FGD1型(OMIM 202200);由MC2R辅助蛋白质(melanocortin 2 receptor accessory protein, MRAP)基因致病变异引起的FGD2型(OMIM 607398);由非典型的类固醇生成急性调节蛋白(steroidogenic acute regulatory protein, STAR)基因致病变异引起的FGD3型。其他报道与FGD相关的基因包括MCM4、NNT、TXNRD2、CYP11A1和SGPL1基因[1-2]。不同病因引起的FGD有不同的临床表现,共同的临床表现为糖皮质激素缺乏。FGD1占FGD的25%,主要表现为皮肤色素沉着、低血糖、反复感染、高胆红素血症和高身材[2-3],也有一些面容异常[3-5],如前额突出、眼间距过宽、内眦赘皮、宽鼻梁和其他部位畸形的报道,如小而尖的手指、通贯掌。本研究报告1例FGD1型病例,具体如下。
1 病例介绍
1.1 临床资料 患儿,女,出生后1 d出现发绀、气促,心率及血氧下降,予胸外心脏按压、气囊加压给氧治疗后心率恢复,血氧欠佳,气管插管下转诊笔者医院。患儿系G1P1孕41周行剖宫产娩出,出生体质量2 920 g,身长50 cm,出生后1、5和10 min Apgar 评分分别为9、10和10分。父母体健,非近亲婚配,家族中无类似症状及遗传性疾病病史。体格检查:血压48/31 mmHg(1 mmHg=133.3 Pa),神志清,全身皮肤黏膜及外阴色素沉着,前额突出,眼距宽,甲状腺无肿大,心、肺、腹未见明显异常,双手通贯掌,阴蒂无肥大。实验室检查肾功能、心肌酶、血脂均正常,总胆红素152.8 μmol/L;血气分析:pH值为7.201、BE为-10.4 mmol/L、碳酸氢根17.5 mmol/L、钾3.84 mmol/L、钠130.4 mmol/L、葡萄糖2.01 mmol/L;血串联质谱及尿有机酸分析未见明显异常;胸部正位片:呼吸窘迫综合征;心脏彩超及颅脑MRI显示未见明显异常;染色体46,XX。入院后,患儿反复出现顽固性低血压(35/20 mmHg),予补液,补充白蛋白,多巴胺、多巴酚丁胺联合去甲肾上腺素升血压处理,血压仍低,加用氢化可的松维持血压,血压稳定后停用,3 d后出现低钠低氯血症(钠133.3 mmol/L,氯97.4 mmol/L)伴高钾血症(钾6.0 mmol/L)。甲状腺功能检查显示,促甲状腺激素(thyroid stimulating hormone, TSH)为25.54 μIU/mL,FT3和FT4正常;ACTH为1 250 pg/mL;皮质醇<0.8 μg/dL,睾酮<7 ng/dL;孕酮<0.21 ng/mL;17α羟孕酮为0.35 nmol/L,雄烯二酮<0.3 ng/mL。甲状腺及肾上腺彩超正常,提示原发性肾上腺功能不全(primary adrenocortical insufficiency, PAI)。加用氢化可的松[27 mg/(m2·d)]及左甲状腺素治疗。患儿PAI诊断成立,建议完善基因检测,明确病因。
1.2 医学外显子组测序结果及分析 在签署知情同意书后,采集患儿及其父母的外周血样2 mL(EDTA抗凝),送厦门基源医学检验实验室进行全外显子组基因检测。从先证者外周血白细胞中提取全基因组DNA,进行DNA建库和捕获,并进行150 bp 双末端测序。测序原始数据经生物信息分析处理后,采用Burrows-Wheeler Alignment tool (BWA)软件进行序列比对,并用GATK软件(https://software.broadinstitute.org/gatk/)分析变异。采用Annovar软件进行变异注释(注释信息包括染色体起始和终止位置,参考等位基因,替代等位基因,基因功能,千人基因组和ExAC等数据库人群频率SIFT/Provean/ Mutation taster等蛋白功能预测软件结果)。结合先证者的临床表型,参考美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准与指南[6-7]对检出变异进行解读,结果显示患儿MC2R基因2号外显子存在2个复合杂合变异:NM_001291911(MC2R):c.145G>A(p.Val49Met),NM_001291911(MC2R):c.536C>A(p.Thr179Lys)(图1),分别遗传自父母。其中c.145G>A(p.Val49Met)变异导致第49位氨基酸由缬氨酸变为甲硫氨酸。该变异在千人基因组数据库中未收录,在ExAC等数据库中等位基因频率为0.000 1,在In-House数据库MAF频率为0.000 014 6,生物信息学软件预测为有害(SIFT: Damaging, Polyphen2_HDIV:possibly damaging, Polyphen2_HVAR: Possibly damaging, Mutation taster: disease_causing),蛋白结构预测可能有害(Polyphen2, SIFT, Mutation taster),软件预测该核苷酸具有高度保守性(GERP, phyloP, phastCons)。根据ACMG指南,判定该变异为可能致病性变异(致病性证据等级为PM1-PM3-PM2_supporting-PP3)。c.536C>A(p.Thr179Lys)变异导致第179位氨基酸由苏氨酸变为赖氨酸。该变异在千人基因组、ExAC、In-House等数据库中未收录,生物信息学软件预测为有害(SIFT: damaging; Polyphen2_HDIV: probably damaging; Polyphen2_HVAR: probably damaging; Mutation taster: disease_causing; Provean: deleterious),蛋白结构预测可能有害(Provean, SIFT, Mutation taster),软件预测该核苷酸具有高度保守性(GERP, phyloP, phastCons)。根据ACMG指南判定该变异为可能致病性变异(致病性证据等级为PM1-PM2_supporting-PM3-PP3)。
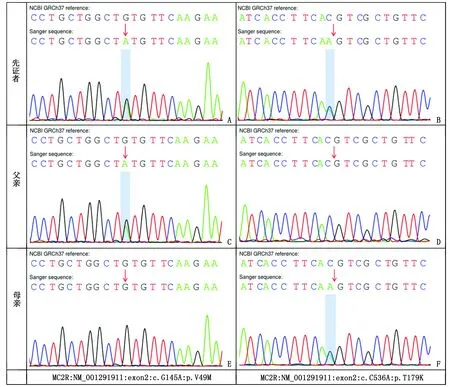
A、B:患儿分别携带c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)杂合变异;C、D:患儿父亲携带c.145G>A(p.Val49Met)杂合变异,未携带c.536C>A(p.Thr179Lys)杂合变异;E、F:患儿母亲未携带c.145G>A(p.Val49Met)杂合变异,携带c.536C>A(p.Thr179Lys)杂合变异。图1 先证者及其父母MC2R基因测序结果Fig.1 MC2R gene sequencing results of the proband and her parents
1.3 随访情况 出院后门诊随诊,监测电解质正常,ACTH 1 250 pg/mL,皮质醇<0.8 μg/dL,予氢化可的松逐渐减至生理需要量。甲状腺功能及神经系统发育正常。尝试停用左甲状腺素,停药2周后,TSH升至6.85 μIU/mL,1个月时升至9.344 μIU/mL,考虑TSH进行性升高,再次予左甲状腺素治疗。现1岁5个月,身长85 cm,体质量10.4 kg,头围45.5 cm,胰岛素样生长因子1(insulin-like growth factor 1, IGF-1)、胰岛素样生长因子结合蛋白(insulin-like growth factor binding protein 3,IGFBP-3)正常,立位醛固酮为275.17 pg/mL,肾素活性为22.15 ng/(mL·h)。
2 讨 论
本研究报道了1例MC2R基因致病变异所致的FGD1新生儿,有助于提高临床医师对FGD1的认识。该患儿出生后即出现皮肤黏膜色素沉着,顽固性低血压,伴有低钠、高钾血症,皮质醇降低,ACTH显著升高,符合PAI的诊断。体见轻度的面部异常,表现为前额突出、眼距宽,伴有双手通贯掌,实验室检查存在甲状腺功能异常,予小剂量左甲状腺素治疗,甲状腺功能维持正常,曾尝试停左甲状腺素,停药后TSH进行性升高,再次予左甲状腺素治疗。门诊随诊监测身长,存在生长加速现象。
FGD1是PAI的一种特殊类型,较罕见,发病率不明确。我国于2019年报道首例FGD1,至今共报道3例[4,8-9]。典型的临床表现为色素沉着、身材高大。随着报道病例的增加,FGD1的临床表型也不断增多。FGD1可能伴有其他的内分泌异常[4-5,10-15],最常见的是甲状腺功能异常[2,4,10,16],多表现为暂时性单纯TSH升高,予左甲状腺素短期治疗或未经治疗后恢复正常。部分FGD1患者存在部分或完全性激素缺乏[4,11,13],如17α羟孕酮、雄烯二酮、脱氢表雄酮、睾酮、孕酮和硫酸脱氢表雄酮。FGD1患者可能出现阴毛发育延迟,但其他性特征不受影响。FGD1临床表现为单纯糖皮质激素缺乏而盐皮质激素活性正常,可见短暂的盐丢失或低钠血症,但真正的盐皮质激素缺乏非常少见[12,14-15]。CHAN等[14]报道6例FGD1出现盐皮质激素不足,均为纯合无义或移码变异,提示MC2R基因严重或纯合变异可干扰肾素-血管紧张素-醛固酮系统,可能需要短暂的盐皮质激素治疗,但不会出现长期的盐皮质激素缺乏。另外,典型的异常面容有助于FGD1的诊断,患者可有面容异常或其他部位畸形。SLAVOTINEK等[5]首次报道1例具有R146H纯合变异的FGD1患儿具有宽鼻梁、内眦赘皮和小而尖的手指。后续研究也报道患儿具有前额突出、眼间距过宽、内眦赘皮或通贯掌等异常面容[3-4];部分合并肌无力[10]、运动发育迟缓[17]等。
本研究患儿查体有前额突出、眼距宽、双手通贯掌,与文献[3-4]报道的病例面容相似。实验室检查17α羟孕酮、雄烯二酮降低,结合患儿月龄处于小青春期,睾酮、孕酮相对偏低,提示存在性激素缺乏。病程中有低钠血症,予糖皮质激素替代,监测电解质正常,查醛固酮水平正常,未提示盐皮质激素缺乏,与上述文献报道一致。本研究患儿已1岁5个月,仍需左甲状腺素治疗,尚需进一步随诊以明确甲状腺功能异常是否也为暂时性。
FGD1出生时身长正常,诊断时身材高大[11-12,18],提示生长加速发生在出生后。本研究中患儿出生时身长正常,新生儿期即予生理替代量的氢化可的松治疗,随访至1岁5个月,身长位于同年龄同性别1.5个标准差,提示在生理替代量氧化可的松治疗的情况下,患儿仍存在生长加速现象。IGF-1和IGFBP-3 水平正常,提示生长加速与生长激素/IGF-1轴无关,与文献报道一致[3,5,13]。有学者认为,FGD1患儿身材高大与长时间暴露于ACTH和慢性糖皮质激素缺乏有关[18],认为FGD1患儿经糖皮质激素替代治疗,ACTH显著下降,晚期生长速率正常化[3,19]。文献中使用的糖皮激素剂量较大,身高正常化可能与大量的糖皮质激素抑制生长相关。也有学者认为,高身材可能是ACTH激活骨和软骨生长板中表达的其他黑皮质素受体并刺激生长,或者刺激雌激素的合成促进生长[20]。
FGD1呈常染色体隐性遗传,本例患儿父母表型正常,Sanger测序验证患儿检出的2个MC2R基因致病变异分别遗传自父母,呈反式排列,与FGD1的遗传模式相符合。
MC2R是7个跨膜的G蛋白偶联受体,在肾上腺皮质高表达,与ACTH结合导致细胞内环磷酸腺苷增加和蛋白激酶A激活,控制着糖皮质激素的分泌[7]。MC2R基因突变导致FGD1的潜在机制可能是由于受体从内质网到细胞表面转运障碍,细胞表面受体表达减少,少数转运至细胞表面的受体位于MC2R的胞外结构域,导致配体与受体结合失败,引起糖皮质激素分泌缺陷[21-22]。本研究中,c.145G>A(p.Val49Met)变异在千人基因组数据库中未检出,在ExAC数据库中等位基因频率极低,采用SIFT、Polyphen2_HDIV、Polyphen2_HVAR、Mutation taster等生物信息学软件预测为对蛋白功能有害的变异,该变异曾在1个FGD1男孩中检出。p.Val49Met变异位于第1个跨膜结构域和第1个胞内环的交界处。根据分子模型,Val49Met变异导致第1个跨膜结构域的结构改变,以及第一跨膜结构域和第7个跨膜结构域潜在新的相互作用,可能导致信号转导丢失,进而导致糖皮质激素分泌缺陷,且缬氨酸在不同物种之间高度保守。在60个不相关的健康对照者中未发现该变异,也提示该变异可能在MC2R功能中发挥重要作用[10]。c.536C>A(p.Thr179Lys)变异在千人基因组、ExAC等数据库中均未检出,采用SIFT、Polyphen2_HDIV、Polyphen2_HVAR、Mutation taster等生物信息学软件预测为对蛋白功能有害的变异,蛋白结构预测可能有害(Polyphen2, SIFT, Mutation taster)。患儿临床表型及遗传方式均与FGD1相符,根据ACMG指南,c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)均为可能致病性变异。因此,认为本例携带的MC2R基因c.145G>A(p.Val49Met)和c.536C>A(p.Thr179Lys)变异是致病变异,尚需进一步的功能研究来验证此变异对MC2R蛋白功能的影响。
综上所述,本研究报道1例新生儿发病的FGD1病例,具有特殊的面部特征,伴甲状腺功能异常;新发现一个MC2R基因致病变异——c.536C>A(p.Thr179Lys),丰富了MC2R基因变异谱。
猜你喜欢
世界最新医学信息文摘(2022年42期)2022-10-28
家庭医药(2022年5期)2022-05-18
中国典型病例大全(2022年7期)2022-04-22
医学概论(2021年18期)2021-01-21
医学概论(2021年18期)2021-01-21
康颐(2020年10期)2020-11-01
支部建设(2020年15期)2020-07-08
科学生活(2016年8期)2016-08-30
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
