
分散液液微萃取/HPLC-MS/MS测定保健茶中非甾体抗炎药
2022-06-13 06:47周斌孙亮张书玲
食品工业 2022年5期
周斌,孙亮*,张书玲
1.杭州市食品药品检验研究院(杭州 310022);2.通标标准技术服务有限公司杭州分公司(杭州 310052)
近年来,不法分子利益驱使,向保健茶中非法添加非甾体抗炎药(NSAIDs)时有发生[1-2]。《中华人民共和国食品安全法》第十条规定,食品中不得加入药物。NSAIDs是世界上使用最广泛的药物,物种类繁多。非甾体抗炎药具有消炎、抗风湿作用,主要有:甲酸类,代表药物二氟尼柳等;乙酸类,代表药物有双氯芬酸、吲哚美辛、舒林酸等;丙酸类,代表药物有芬布芬、萘普生、奥沙普秦等;昔康类,代表药物包括吡罗昔康、美洛昔康等;昔布类,代表药物包括塞来昔布[3]。非甾体抗炎药会引起肠胃疾病、肝肾毒害、心肌梗塞、中风等[4-6]。长期食用非法添加药物,严重危害食用者健康[7]。
目前非法添加主要检测方法有毛细管电泳法[8]、高效液相色谱法[9-10]、高效液相色谱-液质联用法[11-14]、电化学检测法[15]、光谱检测法[16-18]。样品前处理常用固相萃取对保健茶中的非法添加NSAIDs进行提取和净化,这种方法耗费大量的有机试剂,耗材和装置昂贵。分散液液微萃取技术是通过形成稳定的萃取剂/分散剂/样品溶液乳浊液体系,使待测物在样品溶液及萃取剂之间快速达到分配平衡而完成萃取[19-20]。其因操作简便、消耗溶剂量少、富集倍数高等优点,己成为国内外研究药物残留分析研究的热点之一。研究采用分散液液微萃取技术与高效液相色谱-串联质谱技术相结合,建立了保健茶中非法添加9种非甾体抗炎药的测定方法,为保健茶中非甾体抗炎药定性和定量检测提供了可靠的分析手段。
1 材料与方法
1.1 材料与试剂
保健茶(市售);美洛昔康、芬布芬、奥沙普秦、萘丁美酮、非普拉宗、塞来昔布(纯度≥99.4%,购自中国食品药品检定研究院);贝诺酯(购自LGC,纯度≥98.9%);吡罗昔康、依托考昔(购自麦克林,纯度≥98%)。以上对照品均供含量测定用。
甲醇、乙腈、甲酸(色谱纯,美国Fisher公司);超纯水(实验室自制)。
1.2 仪器与设备
AB 4000 QTRAP型串联三重四极杆质谱(美国AB SCIEX公司);Milli-Q超纯水仪(美国Millipore公司)。
1.3 方法
1.3.1 标准溶液配制
标准储备液(500 μg/mL):准确称取标准品各50.0 mg(精确至0.000 1 g),分别置于100 mL容量瓶中,美洛昔康加5 mL水溶解,再用甲醇稀释至刻度,其余标准品用甲醇溶解并稀释至刻度,摇匀,配制成质量浓度各为500 μg/mL标准储备液,于-20 ℃保存备用。
1.3.2 样品前处理
称取1.00 g样品置于15 mL聚丙烯离心管中,加入10 mL乙腈,按4 500 r/min离心5 min,吸取1.0 mL乙腈提取液,加入5.00 mL水和50 μL四氯化碳,涡旋提取3 min,按7 000 r/min离心10 min,用微量注射器吸取底部一定量的沉淀相,氮气吹干,用甲醇溶解定容至0.5 mL,过0.22 μm滤膜,供LC-MS/MS测定。
1.3.3 色谱条件
色谱柱:Acquity BEH-C18柱(2.1 mm×100 mm,1.6 μm);流速:0.4 mL/min。柱温:35 ℃;进样量:5 μL。流动相:B为乙腈,A为含0.05%甲酸的2 mmol/L乙酸铵溶液。梯度洗脱程序:0~1 min,5%B;1.1~5 min,5% B~45% B;5.1~9 min,45% B;9.1~10 min,45% B~55% B;10.1~11 min,55% B,11.1~12 min,95% B,12.1~13 min,5% B。
1.3.4 质谱条件
质谱离子源为ESI+;扫描方式:多反应监测(MRM);去溶剂温度(TEM):500 ℃;气帘气压力(CUR):20 psi;碰撞气压力(CAD):10 psi;离子气1压力(GS1):55 psi;离子气2压力(GS2):60 psi;离子喷射电压(IS):5 500 V(正离子模式)。
采用MRM-信息依懒性采集(IDA)-EPI模式,建立EPI图谱库;MS条件下添加信息相关扫描,监测响应值超过1 000化合物,得到其离子信息,扫描范围m/z50~500,去簇电压60 V,碰撞能量35±15 eV;扩展碰撞能量15 V;一级全扫描获得的高精度相对分子质量数据用于定量及定性分析,IDA全扫描获得的二级碎片离子信息用于加强定性确证。
化合物定性、定量离子及其他质谱参数见表1。
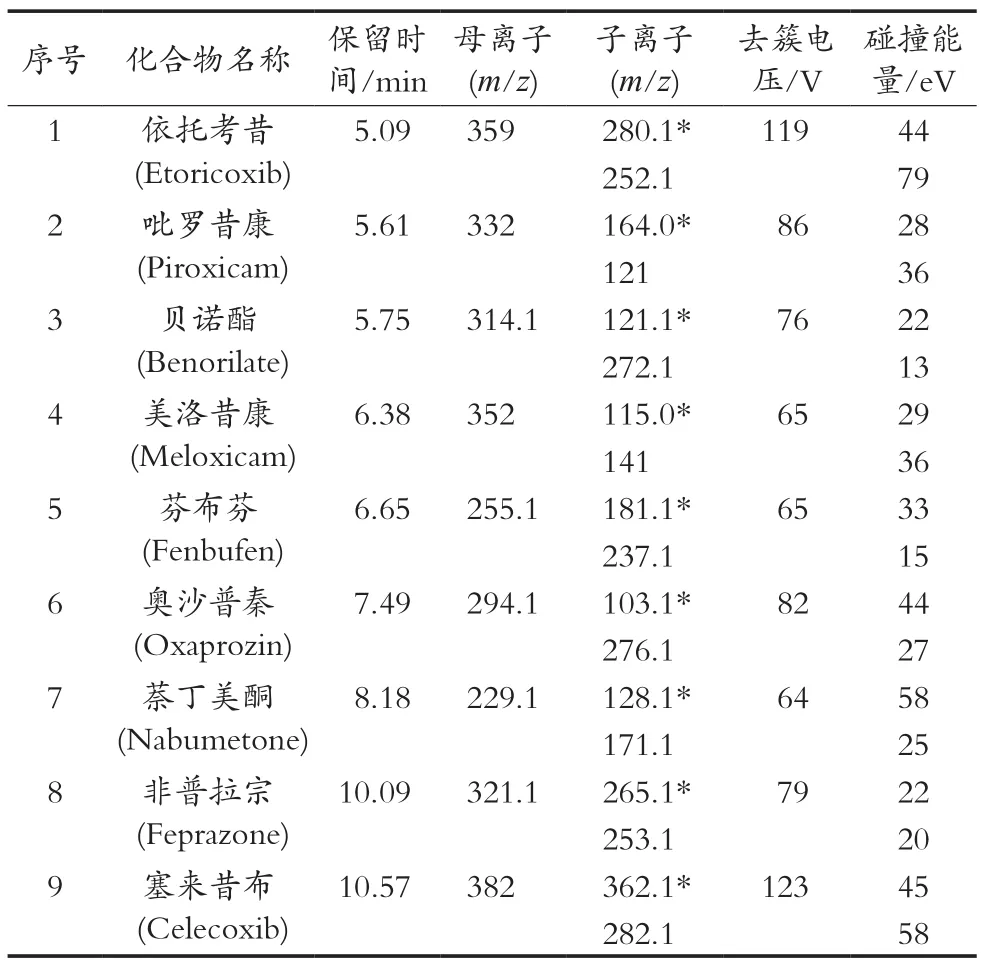
表1 9种非甾体抗炎药质谱参数
2 结果与分析
2.1 流动相的选择
试验比较研究了在乙腈-水流动相体系中分别加入0.1%甲酸水-乙腈、0.05%甲酸水-乙腈、5 mmol/L乙酸铵、2 mmol/L乙酸铵、5 mmol/L乙酸铵(含0.1%甲酸)-乙腈等的影响,并进行了梯度洗脱条件的优化。结果表明,以0.1%甲酸的5 mmol/L乙酸铵溶液作为流动相,9种化合物的色谱峰形、分离度和质谱信号响应较好。

图1 9种非甾体抗炎药混标总离子流图
2.2 萃取溶剂的选择
萃取剂需与水有较小的溶解能力,密度大于水以便萃取溶剂沉淀,对待测化合物的溶解性能好,以便后续色谱分析。试验选择水作为分散剂,按“2.2”方法进行萃取测定。考察常用的二氯甲烷、氯苯、四氯化碳、四氯乙烯作为萃取剂的萃取效果,其密度分别为1.325,1.11,1.59和1.62 g/mL。图2表明:二氯甲烷作为萃取剂的结果重现性较差,可能是因为二氯化碳易挥发,导致萃取液体积变化大;四氯化碳的萃取效率(90.0%~104.1%)比氯苯(84.6%~93.1%)和四氯乙烯(77.9%~92.1%)要好,四氯化碳的萃取效率最好,故选择四氯化碳为目标化合物的萃取溶剂。
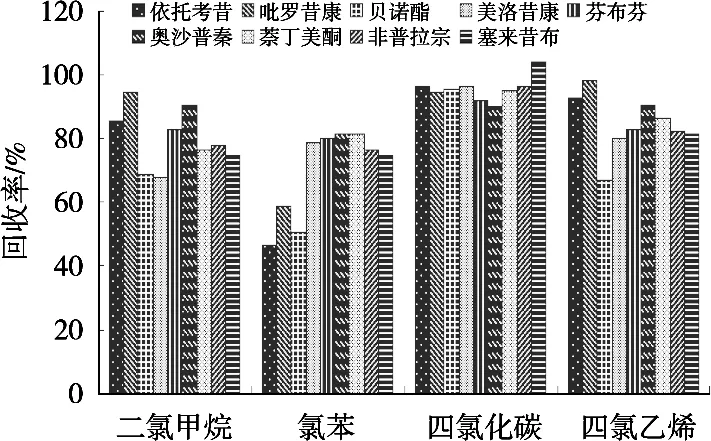
图2 萃取剂种类对回收率的影响
2.3 提取溶剂的优化
试验比较了在100 μg/kg浓度下,甲醇、乙腈、丙酮、乙酸乙酯对9种非甾体抗炎药萃取效果试验。图3表明,采用乙腈作为提取溶剂时的效果最高,回收率能达到89.5%~110%,而且不易提取色素,杂质干扰少。

图3 在100 μg/kg浓度下9种非甾体抗炎药在不同提取溶剂下的回收率
2.4 萃取条件的优化
试验通过考察萃取剂体积、分散剂体积、水的体积对萃取效率的影响,以测定美洛昔康的回收率的影响,确定最优萃取效率。
2.4.1 分散剂体积对回收率的影响
试验通过考察不同体积(30,50和90 μL)的萃取剂四氯化碳对美洛昔康回收率的影响。结果表明,萃取剂为50 μL时,美洛昔康回收率回收率最高,因此萃取剂的体积选择50 μL。
2.4.2 分散剂体积对回收率的影响
考察不同分散剂体积(0.5,1.0和1.5 mL)对美洛昔康回收率的影响。结果表明,分散剂体积为1.0 mL时,美洛昔康回收率最高。结果表明,分散剂体积直接影响待测物在水中的溶解度,合适体积的分散剂不仅能提高传质效果,还能提高萃取效率。
2.4.3 水的体积对回收率的影响
考察不同水的体积(2.5,5.0和7.5 mL)对美洛昔康的回收率影响。结果表明,水的体积为5 mL时,美洛昔康的回收率最高。可能是因为水的体积大于5 mL时,部分萃取剂会溶解在水中,导致萃取效率降低。因此,最优萃取条件中水的用量为5 mL。
2.5 方法学验证(标准曲线、线性范围、检出限、定量限、相关系数)
取混合标准工作液,用甲醇配制成系列标准溶液,以色谱峰面积为纵坐标,各组分质量浓度为横坐标,分别绘制标准曲线,并计算回归方程、线性范围以及相关系数。在相应的质量浓度范围内,9种化合物的线性关系良好,相关系数r均在0.998以上。
取阴性保健茶,添加不同浓度的待测物,以性噪比(S/N)=3计算方法检出限,S/N=10计算方法定量限,检出限为0.5~5 μg/kg,定量限为1.5~15 μg/kg。
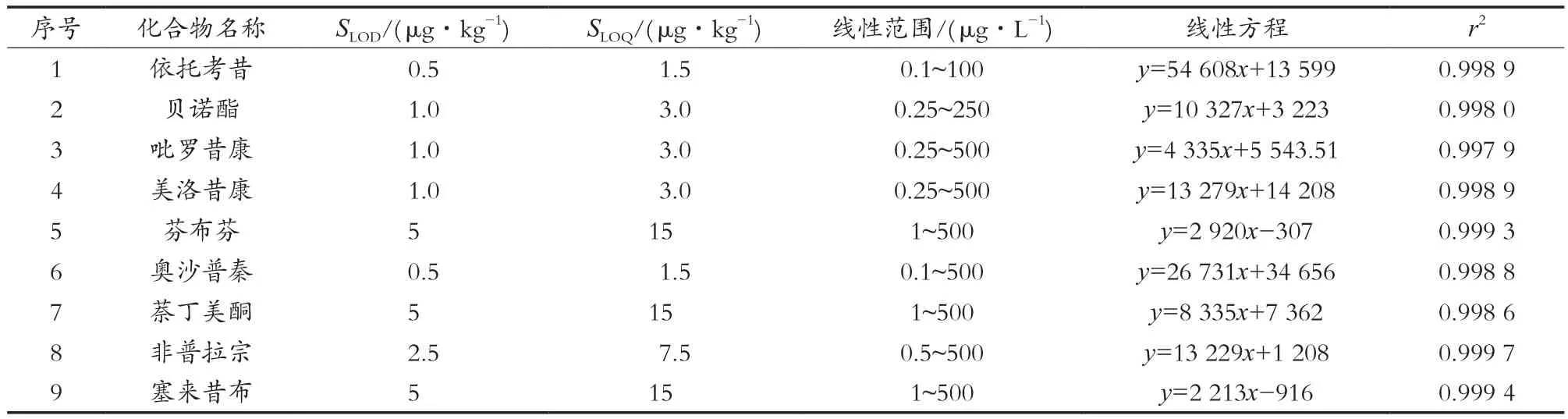
表2 9种非甾体抗炎药的检出限、定量限、线性范围、线性方程、相关系数(r)
2.6 方法的添加回收率、精密度
取6份不含本底的保健茶,每份1.00 g,分别添加0.02,0.05和0.2 mg/kg 3个水平质量浓度的混合标准储备液。按2.2方法测定回收率,每个水平测定5次,结果见表4。结果显示,在0.02,0.05和0.2 mg/kg 3个添加水平下,平均回收率为85.7%~105.7%,相对标准偏差(SRSD)为1.7%~8.9%,满足定量分析的要求。

表4 阳性样品EPI谱库搜索匹配结果
2.7 实际样品分析
用该方法测定了从网上购买的56批次保健茶。检出一个批次进口保健茶中含有吡罗昔康,EPI谱库搜索匹配度≥90,为阳性样品,含量为7.42 g/kg。
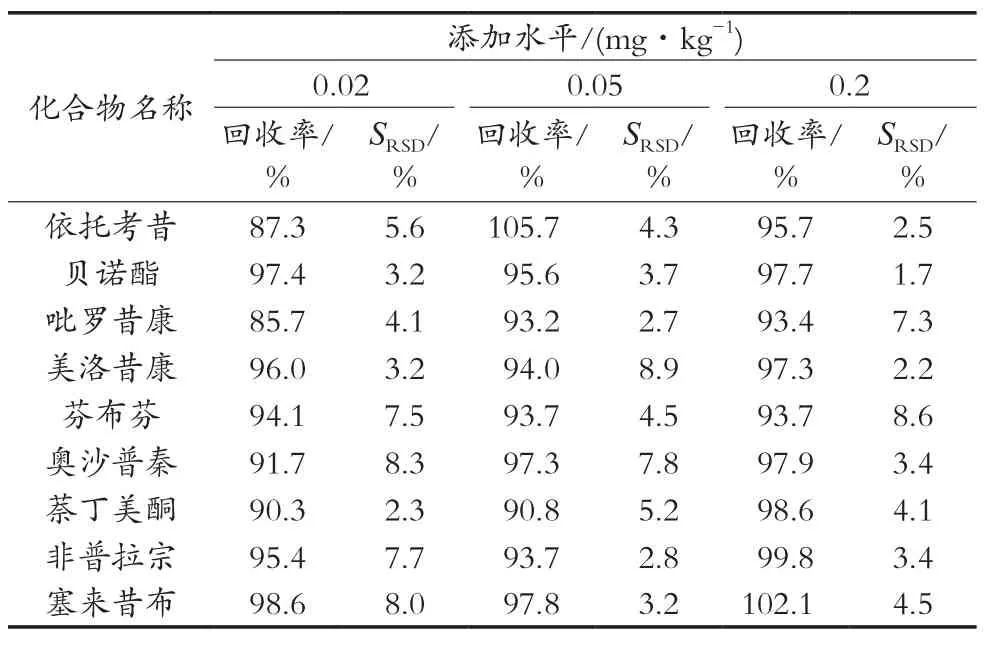
表3 9种非甾体抗炎药的添加回收率和精密度(n=6)

图4 阳性样品和标准溶液(50 μg/L)增强离子扫描(EPI)图谱
3 结论
将分散液液微萃取技术与液相色谱-串联质谱法(UPLC-MS/MS)相结合应用于保健茶中9种非甾体抗炎药的检测。与常规保健食品中非法添加非甾体抗炎药的液相质谱法相比,该方法操作简单,选择性好,灵敏度高,没有繁琐的净化过程,有效地避免假阳性样品。该方法可以应用于种类繁多和样品量巨大的保健茶中非法添加非甾体抗炎药快速筛查,为相关的监管部门提供了有效的技术支持。
猜你喜欢
中国化工贸易·下旬刊(2019年10期)2019-10-21
中国保健营养(2019年6期)2019-10-21
科学导报(2019年19期)2019-09-23
农家科技(2019年1期)2019-03-13
家庭用药(2017年7期)2017-07-29
医学美学美容·中旬刊(2015年2期)2015-10-21
中国信息化·学术版(2013年3期)2013-06-25
