
盐酸屈他维林的晶体结构及其含量测定方法
2022-07-01 06:20张雍洁谭忠琴
武汉工程大学学报 2022年3期
赵 芳,张雍洁,谭忠琴,王 凯*
1.湖北大学化学化工学院,湖北 武汉 430062;
2.上海信息技术学校,上海 200331
盐酸屈他维林,化学名为1-(3,4- diethoxybenzyl) -6,7-diethoxy-3,4-dihydroiso-quinoline hydrochloride,为匈牙利Chinoin 公司所研制,并于20 世纪60 年代被应用于临床中,20 世纪90 年代开始在中国推广[1]。在病理情况下,内脏或者全身平滑肌异常的剧烈收缩会引起平滑肌痉挛。临床常见的解痉药分为生物碱类M 胆碱受体阻断药、季铵类抗胆碱能药、叔胺类抗胆碱能药以及罂粟碱衍生物,而盐酸屈他维林则属于罂粟碱解痉药类[2-4],因其只作用于平滑肌而不影响自主神经系统,因此适用范围较禁忌青光眼和前列腺肥大的抗胆碱类解痉药更广[5-7];此外,与其他罂粟碱类解痉药相比,盐酸屈他维林的解痉作用更强,而且作用时间更长[8-10]。
不同的药物互变异构体,其理化性质如热稳定性、亲疏水性、极性和颜色等方面会有显著的差异,相应地,在晶体结构、稳定性、可生产性和生物利用度等性质方面也会有所差别[11]。盐酸屈他维林虽是市场前景广阔的解痉药,但对其单晶结构的研究少,结构也存在一定的争议,尤其是双键的位置。药物含量是评价药物质量的主要手段,也是药物质量标准的重要内容[12],其分析方法主要包括容量分析法(滴定法)、光谱分析法和色谱分析法[13-14]。对原料药进行含量测定可为后续药物制剂研究开发、药理毒理研究以及临床试验等的开展奠定基础,然而目前对盐酸屈他维林原料药进行含量测定鲜有报道。因此,认为研究盐酸屈他维林原料药晶体结构并对其进行含量测定是非常必要的。
笔者合成了盐酸屈他维林,通过一系列表征确认其结构,并对其晶体结构进行分析;采用紫外分光光度法和电位滴定法分别建立自制盐酸屈他维林含量测定方法,同时进行了方法学验证。
1 实验部分
1.1 材料、试剂与仪器
样品:自制盐酸屈他维林对照品(质量分数99.5%,批号:S200701,为自制品3 次重结晶);盐酸屈他维林供试品(批号为200715、200722、200729,中国上海麦克林生化科技有限公司)。
试剂:水(超纯水);其他试剂均为分析纯。
仪器:X 射线衍射仪(D8 ADVANCE,中国布鲁克科技有限公司);面探衍射仪(Bruker SMART 1000CDD,中国布鲁克科技有限公司);电子天平(F2014,中国梅特勒公司),UV3110 紫外-可见检测器(EClassical UV P3100,中国西安禾普生物有限公司),紫外-可见光谱仪(TU-1901,中国上海如海光电科技有限公司),电位滴定仪(ZD-2A,中国上海仪电科学仪器股份有限公司)。
1.2 实验方法
1.2.1 自制盐酸屈他维林晶型的测定 (1)盐酸屈他维林粉末结晶的制备:称取1 g 自制盐酸屈他维林原料药,通过柱层析纯化,流动相为乙酸乙酯-甲醇(体积比=2∶1),得到精品,再加入3 mL 乙醇-水(体积比=5∶1),加热回流溶解。静止冷却,固体析出,抽滤,烘干,即得盐酸屈他维林淡黄色粉末结晶。采用研钵将其固体颗粒研磨成适当大小的粉末,待用。
(2)盐酸屈他维林单晶制备:采取微量溶剂挥发法。称取0.5 g 经柱层析分离的盐酸屈他维林,加入5 mL 乙醇溶解,放入带有空隙的样品瓶中,静置缓慢挥发溶剂,25 ℃下,放置10 d 后有晶体析出,即得盐酸屈他维林淡黄色片状晶体,待测。
1.2.2 紫外分光光度法含量测定 (1)溶液配制。自制盐酸屈他维林对照品/供试品母液的制备:精密称取10 mg 自制盐酸屈他维林对照品/供试品,置于100 mL 容量瓶中,加甲醇溶解并稀释至刻度,制得0.10 mg/mL 盐酸屈他维林对照品/供试品母液。
(2)标准曲线的制备。利用移液管分别移取0.10 mg/mL 自制盐酸屈他维林对照品母液0.4、0.6、0.8、1、1.2 mL 置于10 mL 容量瓶中,加甲醇稀释至刻度,依次制成4、6、8、10、12 μg/mL 线性曲线供试液。以甲醇为空白溶剂,依次利用紫外分光光度计测量上述系列浓度,以浓度为横坐标,吸收度值为纵坐标,绘制标准曲线。
(3)样品测定。分别精密称取3 个批次自制盐酸屈他维林供试品,配制一定浓度的供试液,在最大吸收波长处测定OD 值,代入标准曲线方程,计算自制原料药中盐酸屈他维林的含量。
1.2.3 电位滴定法含量测定 (1)精密称定自制盐酸屈他维林250 mg,加入50 mL 乙醇溶解,再加5 mL 的0.01 mol/L HCl 溶液,打开搅拌功能,使对照品充分溶解,按照电位滴定法,用0.10 mol/L 氢氧化钠滴定液滴定,记录所消耗滴定液体积与电位变化,读取两个拐点所消耗的氢氧化钠滴定液体积之差,消耗1 mL 0.10 mol/L 氢氧化钠滴定液相当于43.40 mg 的C24H31NO4·HCl,同时,按上述操作,设计空白试验对以上自制盐酸屈他维林含量测定结果进行校正。
(2)利用(1)中建立的醇中碱滴定法测定自制盐酸屈他维林的含量。分别精密称取3 个批次自制盐酸屈他维林250 mg,配制一定浓度的供试液并采用电位滴定仪记录,根据氢氧化钠溶液消耗体积计算原料药中盐酸屈他维林的含量。
2 结果与讨论
2.1 自制盐酸屈他维林晶型的鉴定
2.1.1 粉末 X- 射线衍射谱(powder X-ray diffraction,PXRD) 自制盐酸屈他维林的粉末衍射谱如图1 所示,该谱图及特征峰与专利[15]所提到的晶型I基本一致,可能含有0.5 个乙醇分子。
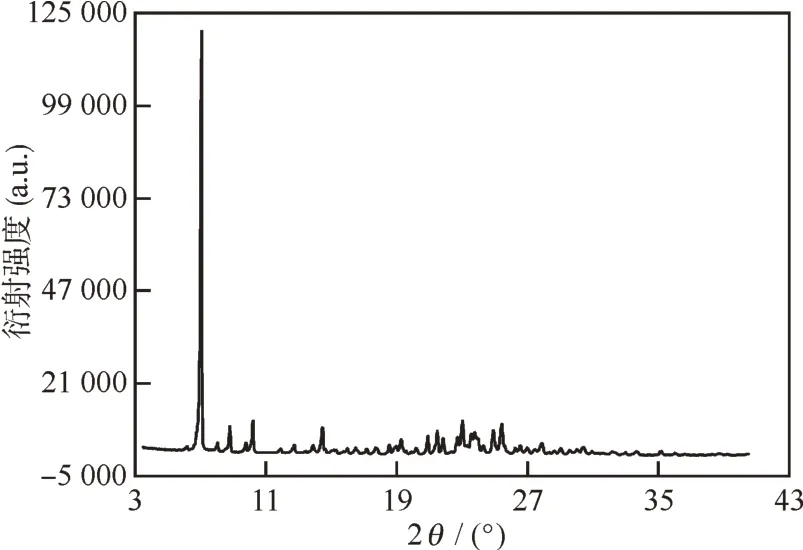
图1 盐酸屈他维林的X-射线粉末衍射光谱Fig.1 Powder X-ray diffraction spectrum of drotaverine hydrochloride
2.1.2 单晶X-射线衍射谱(single-crystal X-ray diffraction,SC-XRD) 对乙醇重结晶得到的盐酸屈他维林晶体进行粉末衍射和单晶X-射线衍射,分别得到晶体结构[图2(a)]和三斜晶系的晶体堆积[图2(b)]。结果表明,盐酸屈他维林属于三斜晶系,P1 空间群,晶体的不对称单元中存在1 个分子(Z’=1),每个晶胞有2 个分子(Z=2)。分析晶体结构,可以发现它是带有0.5 个乙醇结晶的喹啉结构,这与根据PXRD 谱图所推断的结果也是相同的,该结构与专利[15]报道盐酸屈他维林的2 种结构中的结构I 吻合(图3),这也验证了Kuz’mina 提出的结构I 占据主导性的说法[16],并且应该作为含有盐酸屈他维林药品的产品规范文件的默认结构。

图2 盐酸屈他维林:(a)晶体结构,(b)三斜晶系分子堆积图Fig.2 Drotaverine hydrochloride:(a)crystal structure,(b)view of molecular packing for triclinic polymorphs
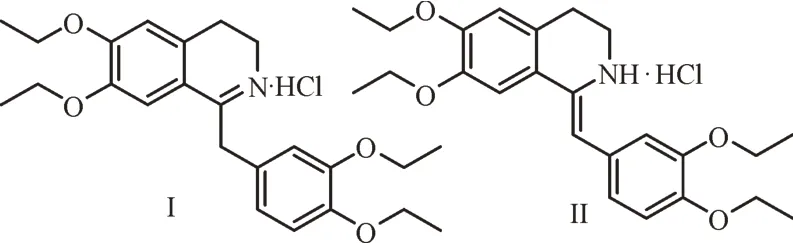
图3 盐酸屈他维林的两种互变异构体Fig.3 Two tautomeric isomers of drotaverine hydrochloride
结果显示,盐酸屈他维林盐酸盐最终结构是含碳氮双键的喹啉结构,并带有0.5 个乙醇结晶。
2.2 紫外分光光度法
2.2.1 检测波长的选择 分别取自制盐酸屈他维林对照品溶液用甲醇适当稀释,以甲醇为空白溶剂,利用紫外分光光度计在200~600 nm 范围内扫描,自制盐酸屈他维林甲醇溶液波长分别为230 、282、305 nm。而230 nm 则为自制盐酸曲维林的最大吸收波长,如图4(a)所示。
通过线性回归建立自制盐酸屈他维林标准曲线方程为A=0.065c+0.008 9,R2=0.999 9,如图4(b)所示,表明盐酸曲他维林在4~12 μg/mL 范围下线性关系良好。

图4 盐酸屈他维林:(a)对照品的紫外可见吸收光谱,(b)样品含量测定的标准曲线Fig.4 Drotaverine hydrochloride:(a)UV-Vis absorption spectrum,(b)standard curve of content determination
2.2.2 精密度试验 取10.00 μg/mL 的自制盐酸屈他维林对照品溶液,利用紫外分光光度计重复测定6 次,计算相对标准偏差(relative standard deviation,RSD)为0.15%,表明仪器的精密度良好。
2.2.3 重复性试验 分别取0.8 mL 自制盐酸屈他维林供供试品母液6 份,置于不同10 mL 容量瓶中,加甲醇稀释至刻度,配制成6 份相同浓度的供试品溶液,利用紫外分光光度计测量,以吸光度值计算RSD 为0.31%,表明样品的重复性良好。
2.2.4 加样回收率试验 取同一批已测得含量为4.00 μg/mL 自制盐酸屈他维林供试品母液9 份各0.4 mL,置于10 mL 容量瓶中,其中3 份为1 组,每组分别加入自制盐酸屈他维林对照品母液0.4、0.5、0.6 mL,加甲醇稀释至刻度,定容,摇匀,得9份回收率供试品溶液。利用紫外分光光度计在230 nm 波长下测量吸光度,按回归曲线方程计算实测值,回收率(%)=(加标实测值-试样测定值)/加标值×100%,算得回收率在98.1%~100%之间浮动,平均回收率为99.14%,RSD=0.59%,表明样品的回收率良好。
2.2.5 稳定性试验 取浓度为8.00 μg/mL 的盐酸屈他维林供试品溶液,室温下放置,于放置时间为0、1、2、4、6、8 h 下测量其吸光度值分别为0.530、0.530、0.530、0.533、0.529、0.530,计算其相对标准偏差,RSD%=0.25%,表明自制盐酸屈他维林在0~8 h 内稳定。
2.2.6 样品含量测定 在最大吸收波长230 nm 处测定OD 值,带入标准曲线方程计算得200715、200722、200729 这3 个批次的样品含量分别为100.15%、101.56%、99.74%。
2.3 电位滴定法
2.3.1 滴定法的选择 结构中含有“胺基”的药物的含量测定通常采用高氯酸滴定法和氢氧化钠(或氢氧化钾)滴定法。高氯酸法和氢氧化钠法分别滴定药物的胺基部分与酸根部分[17-18]。一方面由于自制盐酸屈他维林中可能存在胺基部分基团的杂质,高氯酸同样会与此类杂质作用,会引起测定误差,另一方面,高氯酸法测量氢卤酸类药物时,氢卤酸会对滴定产生干扰,需添加醋酸汞抗干扰[19];而氢氧化钠滴定法,所用试剂均为常用试剂,对环境影响小,且不需要严格控制周围环境,故考虑使用氢氧化钠滴定法测定盐酸曲他维林含量。本文采用乙醇为测量溶剂,该法又叫醇中碱滴定法[20-21]。
2.3.2 含量测定 使用电位滴定法对自制盐酸屈他维林进行含量测定,以一级微商曲线确定终点,其滴定曲线如图5(a)所示。
2.3.3 线性关系考察 分别精密称定同一批自制盐酸屈他维林100、150、200、250、300 mg,相当于250 mg 的40%、60%、80%、100%、120%。记录各样品所消耗氢氧化钠体积,两个拐点所对应体积之差与对应称量样品重量用最小二乘法进行线性回归分析,结果显示[图5(b)],样品范围在0.1~0.3 g范围内线性方程为y=25.45x-0.151 3,R2=0.997 6,说明该方法具有可行性。
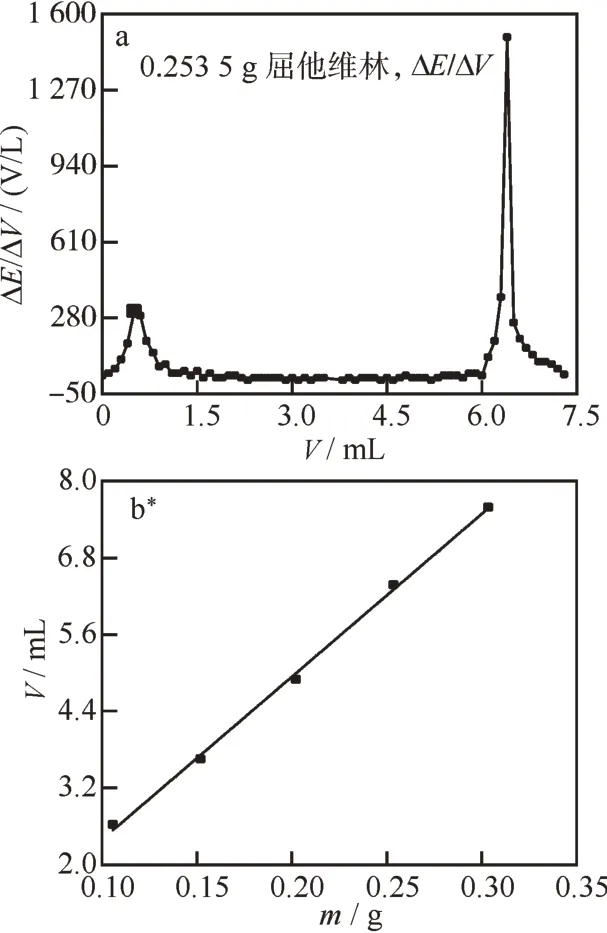
图5 盐酸屈他维林:(a)电位滴定曲线,(b)含量测定标准曲线Fig.5 Drotaverine hydrochloride:(a)potentiometric titration curve,(b)standard curve of content determination
2.3.4 重复性实验 分别精密称定同一批自制盐酸屈他维林100 mg,共6 份,按《方法与结果》测定,记录电位变化及氢氧化钠消耗体积,计算各样品含量为99.12%~100.02% 不等,平均含量为99.69%,RSD 为0.47%,表明该方法重复性良好。
2.3.5 回收率实验 分别精密称定盐酸屈他维林原料药对照品200、250、300 mg,各3 份,共9 份,按《方法与结果》测定并计算平均回收率为99.9%,RSD 为0.29%,结果表明,该方法回收率良好。
2.3.6 样品含量测定 结果表明3 批自制盐酸屈他 维 林 含 量(200715、200722、200729)分 别 为99.51%、101.87%、100.03%,与紫外分光光度法所测结果基本一致,且符合原料药质量研究。
2.4 方法比较
紫外分光光度法样品用量少,但受具有相似吸收波长杂质的影响,不易控制。电位滴定法避免了传统剧毒物质的使用且不受杂质影响,虽样品用量大,但对于每次样品含量测定与其他两种测定方法相比减少了标准曲线方程的制备,可直接滴定即可获得含量,节省了时间。
3 结 论
笔者自制了盐酸屈他维林并通过PXRD 和SCXRD 对分子进行结构确认,确认了其晶型是含有0.5 个乙醇分子的喹啉结构,且属于C=N 互变异构,与文献报道结构一致[16],对盐酸屈他维林晶体结构的研究为后续对其进行药物多晶型研究提供了基础。通过采用紫外分光光度法和电位滴定法建立自制盐酸屈他维林含量测定的方法,并进行方法学验证,验证结果表明:二种方法均具有良好的可行性,使用该方法对自制盐酸屈他维林供试品进行含量测定,实验结果证明了自制盐酸屈他维林符合原料药质量研究,为后续新药申报奠定了基础。
猜你喜欢
土壤学报(2022年3期)2022-08-27
全面腐蚀控制(2022年2期)2022-03-11
中国听力语言康复科学杂志(2021年6期)2021-12-21
初中生学习指导·中考版(2021年2期)2021-09-10
世界家苑(2018年6期)2018-07-23
神州·下旬刊(2017年6期)2017-10-28
中学生数理化·中考版(2015年12期)2015-09-10
数理化学习·教育理论版(2013年9期)2013-12-27
中国信息化·学术版(2013年5期)2013-10-09
数理化学习·初中版(2009年3期)2009-04-21
