
由2 例罕见病引发的临床思考
2022-07-06 23:43姜秀芳李娟王婷袁兆红
中国典型病例大全 2022年17期
姜秀芳 李娟 王婷 袁兆红


【中图分类号】 R4【文献标识码】A 【文章编号】1673-9026(2022)17--03
在儿童保健的常规查体中,在结束就诊时家长经常会问到的问题是“我家孩子发育是否正常?”,由于儿童发育的多样性、多变性、系统性、复杂性,以及临床表现多方面的重叠、交叉或表现不典型,有时临床医生很难用“发育正常还是不正常”这么简单的话来回答家长的问题,在对孩子进行系统的查体时,有时我们会发现一些特殊疾病,这就要求我们临床医生做到早期识别。现分享在查体过程中发现的2例特殊患儿。
一、病例资料
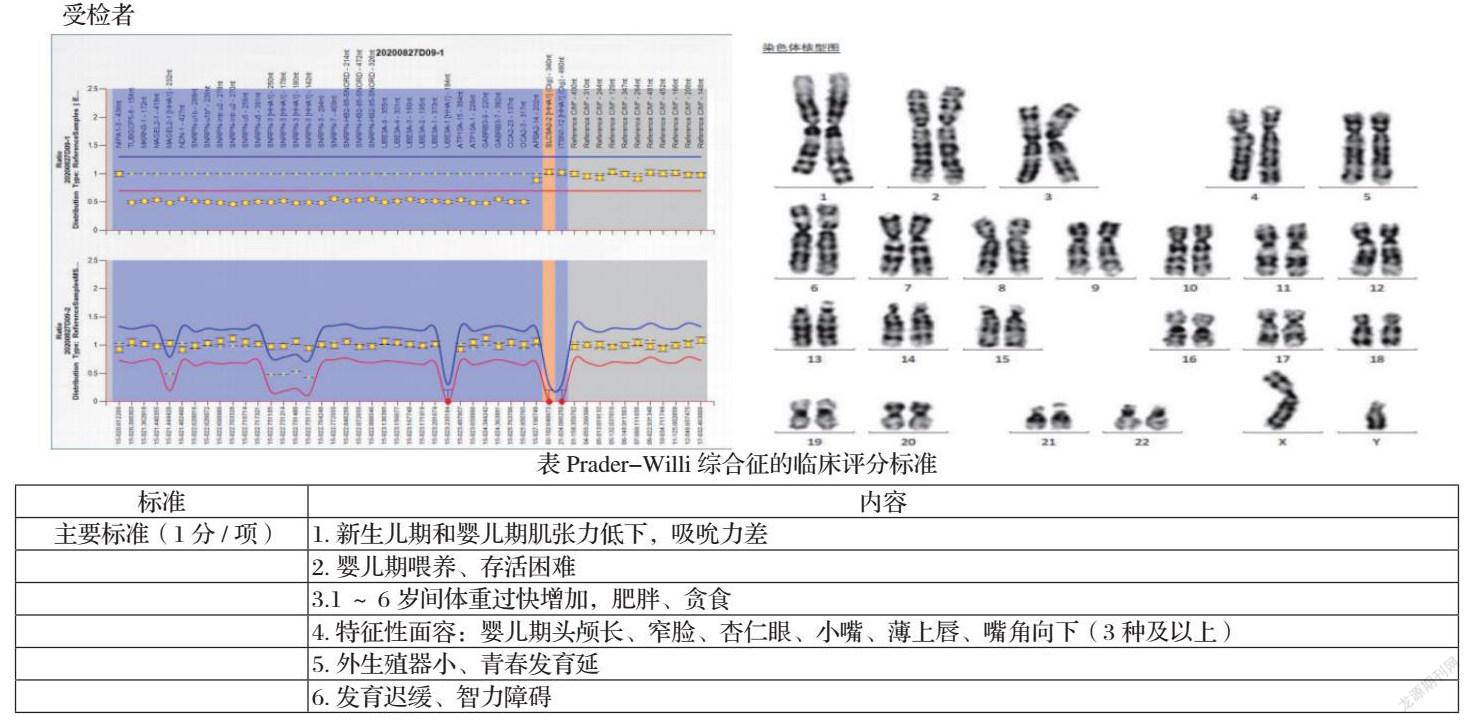
例1:王XX,男,2月14天,于2020年9月18日生后第一次查体。系G1P1,母孕期血糖高,具体不详,羊水少,孕后期胎动少,胎心监护不通过,38+1周因“胎心异常”选择剖宫产,出生体重2.4kg,生后因“胎粪吸入”在NICU住院,具体不详。出院后母乳喂养,吃奶时吸吮无力,吃奶速度慢,每次喂养时间接近1小时,哭声小,少哭闹,反应慢,上肢活动少。新生儿疾病筛查通过。无类似家族史。体格检查:体重3.95kg,身高55.5cm,头围36.5cm,前囟平软。追视反应可引出,重复性欠佳。全身皮肤白皙,稍肿。双肺呼吸音清,未闻及啰音。心音有力,律齐。腹软。阴囊内未触及睾丸。四肢肌张力低,股角、腘窝角均180°,足背屈角45°,围巾征阳性,Moro反射、ATNR引出迟缓,四肢自主活动少,腕下垂,拉起头背屈明显,俯卧位周支撑抬头小于15°,维持时间小于5秒。
例2:董XX,女,39天,于2020年11月26日生后第一次查体。系G1P1,母孕期无异常,足月顺产,出生体重2.85kg,出生身长50.0cm,无缺氧窒息病史,新生儿疾病筛查、听力筛查通过。生后四肢活动少,母乳喂养,吃奶吸吮力及吞咽的协调性尚可,无呛咳,睡眠可,大小便无异常。父母非近亲结婚,家族史阴性。体格检查:体重4.4kg,身长55.0cm,头围35.0cm,前囟1.5×1.5cm,平软。会注视人脸及追视人脸范围达90°,对声音有易惊的动作。无胸廓凹陷。双肺呼吸音清,未闻及落音。心音有力,律齐。腹软。四肢肌张力及肌力低,上肢肌力4级,下肢肌力1级,双侧握持反射均减弱,俯卧位头不能抬离床面。
二、诊断思路
上述2例病例均存在运动功能异常,其中例1除运动功能异常外,还存在吃奶差,皮肤白皙、肿,隐睾,2例患儿均进行基因检查,结果如下:
例1检查结果如下:
检测结果:发现受检者父源性15q11-q13片段缺失变异,染色体核型分析:46,XY。
结果说明:所发现的15q11-q13片段缺失变异为导致受检者发病的致病性变异,父源性15q11-q13片段缺失变异会导致Prader—Willi综合征
根据患儿临床表现,结合检查结果,例1诊断为“Prader—Willi综合征”
Prader—Willi综合征 (PWS)又称肌张力低下一智能障碍一性腺发育滞后一肥胖综合征、普拉德.威利综合征,由Prader等[1]于1956年首次报道,是最早被证实涉及基因组印记的遗传性疾病[2]。国外不同人群的发病率约为1/10 000~1/30 000,我国缺乏流行病学资料[3-5]。PWS是一种15号染色体异常的疾病,在第15号染色体印迹基因区(15q11.2)存在基因缺陷。缺陷基因在大脑所有组织区均有表达,导致了所编码的小RNA(small RNA)缺少和snRNA的修饰异常,进而出现丘脑功能障碍和发育延迟[6]。在疾病基因型中父系染色体基因小片段丢失的概率为65-75%、母系单亲源二体占20-30%、印记缺陷占1-3%[7]。PWS表现为胎儿期胎动少;新生儿肌张力低,哭声弱,喂养困难,婴儿期生长不良,之后出现食欲大增;儿童期因多食导致肥胖症,呈矮胖外观,由于下丘脑发生了病变,患者即使在进食后也没有饱腹感,而生长激素和促性腺激素的缺乏进一步促使肥胖和代谢综合征,认知功能损害参差不齐,运动、语言发育差;青春期以肥胖、性腺发育不良、学习困难为特征[8]。目前国际上通行的PWS临床评分标准主要根据Holm [9] 等于1993年提出、2012年Cassidy等[3]修正后的标准:包括6条主要標准、11条次要标准和8条支持证据。年龄<3岁总评分5分以上,主要诊断标准达4分即可诊断;年龄I>3岁总评分8分以上,主要诊断标准达5分即可诊断(见下表)。PWS是症状性病态肥胖的重要病因之一,早期诊断和合理干预对改善患儿的生活质量、预防严重并发症和延长寿命至关重要[2-5,10,11]。PWS的治疗应采取综合管理模式:饮食行为与营养管理;性腺发育不良及青春期发育问题的处理;生长激素(GH)治疗;、其他内分泌问题的处理[12]。
结果说明:
1.SMN1 基因是脊髓肌萎缩(SMA)的致病基因,为常染色体隐性遗传方式(AR)。
2. SMN2 基因的拷贝数与 SMA 患者临床表现相关,普遍认为:SMN2 基因拷贝数越多,疾病表现相对越轻。
3. 在受检者 SMN1 基因 7、8 号外显子发现的纯合缺失变异有可能是导致受检者发病的致病性变异,受检者其父母有 可能均为致病变异携带者。
根据患儿临床表现,结合检查结果,例2诊断为“脊肌萎缩症”。
脊肌萎缩症(spinal muscular atrophy,SMA)是一种常染色体隐性遗传的神经肌肉疾病,其疾病特点是脊髓运动神经元的变性导致骨骼肌萎缩和普遍性的肌张力减弱[13],多以周围神经肌肉病变为初始症状。SMA按照发病年龄和严重程度的不同,分为4种临床类型,分别是SMA I~IV。SMA 的确诊依赖于基因诊断,主要致病基因是运动神经元基因1(survival motor neuron gene 1,SMN1),同时,SMN2基因(SMN1基因的高度同源基因)[14]和神经原凋亡抑制蛋白基(uronal-apoptosis inhibitory protein,NAIP)也被认为是SMA 疾病重要相关基因[15-16]。SMA在正常出生人群中的发生率大约是1/6000~1/10000,在中国人群中的SMA 相关基因部分缺失携带率为1/42[17]。SMA相关基因的部分缺失也会出现在正常人群中,SMN1基因外显子的部分缺失可以被相关基因功能上弥补,提示SMN1基因缺失致病存在基因剂量效应。同时,这种SMN1基因部分缺失的正常人在后期的生育过程中,可能会拥有SMN基因纯合缺失的后代。因此在SMA 的遗传防治过程中,SMA相关基因筛查具有重要意义[18]。美国FDA 于2016 年12 月23 日批准一种名为Spinraza( nusinersen) 的新药用于治疗儿童及成人SMA ,这也是FDA 批准的第1 种用于治疗该疾病的药物[19]。DB3C5DA0-053D-4195-A1E6-76518BF48BED
三、思考
在儿童保健查体中到底查什么?是不是测量一下身长、体重、头围就可以了?当然没有这么简单。在查体过程中要关注一个主体,一是带养人,另一个是患者。
1.问病史
要询问带养人吃奶情况(母乳/配方奶/辅食添加?吃奶量、吃奶速度、每次喂奶时间及两次奶间隔、吸吮及吞咽情况等)、睡眠情况(过少、过多、晚、哭闹、不规律等)、大小便情况、居住环境、家庭经济状况、有无家族史、传染病、宝宝的预防接种史、孕产情况及生后生长情况、肢体活动情况(过多、过少、运动轨迹欠协调、姿势及肌张力是否稳定等)等。
2.查体
1.先天发育畸形重要脏器的检查、外观检查(排除先天发育畸形);
2.营养状态:身长、体重、头围、上臂围、皮下脂肪等;
3.神经心理发育:观察自主活动情况、与医生和带养人互动情况(视听及玩具的反应)、运动(自主活动、诱导下或被动活动等,能否完成,完成质量如何)、姿势、反射、肌张力、肌力、认知、语言、适应情况等。
在查体过程中要详细,过硬的理论知识及临床经验的积累是避免漏诊、误诊的最有效的保障。在临床工作中,对于第一次来诊的患者,或者随访过程中发现发育进展缓慢或发育倒退的患者一定要引起足够的重视,
参考文献:
[1]Prader A,Labhart A,Willi H.Ein Syndrom von Adipositas,Kleinwuchs, Kryptorchismus and Oligophrenia nachmyatonieartigem Zustand in Neugeborenenalter[J].Schweiz Med Wochenschr,1956,86:1260—1261.
[2]McCandless SE;Committee on Genetics.Clinical report—health supervision for children with Pradcr—Willi syndrome[J].Pediatrics,2011,127(1):195-204.
[3]Cassidy SB,Schwartz S,Miller几,et a1.Prader-Willi syndrome [J].Genet Med,2012,14(1):10-26.
[4]Buffer MG.Prader-Willi syndrome:obesity due to genomic imprinting[J].Curt Genomics,2011,12(3):204-215.
[5]Goldstone AP,Holland AJ,Hauffa BP,et a1.Recommendations for the diagnosis and management of Prader-Willi syndrome[J].JClin Endocrinol Metab,2008,93(11):4183-4197.
[6] Tala D L S, Johannes S, Christian A R, et al . Small Evolutionarily Conserved RNA, Resembling C/D Box Small Nucleolar RNA, Is Transcribed from PWCR1 , a Novel Imprinted Gene in the Prader-Willi Deletion Region, Which Is Highly Expressed in Brain[J]. Am J Hum Genet, 2000, 67(5): 1067-1082.
[7] Yonatan S, Ido S, Ofra Y, et al . The noncoding RNA IPW regulates the imprinted DLK1-DIO3 locus in an induced pluripotent stem cell model of Prader-Willi syndrome[J]. Nat Genet,2014, 46(6): 551-557.
[8]沈春芳,洪洁,叶蕾,等.一例Prader-Willi综合征的临床及表观遗传学研究.中华内分泌代谢杂志,2006,22(5):451-453.
[9]Holm VA,Cassidy SB,Bulter MG,et al. Prader-Willi syndrome:Consensus diagnosis criteria.Pediatrics,1993,91:398-402.
[10]Diene G,Mimoun E,Feigedova E,et a1.Endocrine disorders in children with Prader—Willi syndrome—data from 142 children of the French database[J].Horm Res Paediatr,2010,74(2):121—128.
[11]Emefick JE,Vogt KS.Endocrine manifestations and management of Prader-willi syndrome[J].Int J Pediatr Endocrinol,2013,2013(1):14.
[12]中華华医学会儿科学分会内分泌遗传代谢学组.《中华儿科杂志》编辑委员会. 中国Prader—Willi综合征诊治专家共识(2015)中华儿科杂志,2015,53(6):419-424.
[13]LUNN MR,WANG CH.Spinal muscular atrophy.Lancet,2008,371(9630):2120-2133.
[14]RUGGIU M,MCGOVERN VL,LOTTI F,et al .Arole for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy.Mol Cell Biol,2012.32(1):126-138.
[15]KOTNAI T,SUTOMO R,SASONGKO TH,et al。A novel mutation at the N-terminal of SMN Tudor domain inhibits its interaction with target proteins.J Neurol,2007.254(5):624-630.
[16]DERAKHSHANDEH-PEYKAR P,ESMAILI M,OUSATIASHTIANI Z ,et al.Molecular analysis of the SMN1 and NAIP genes in Iranian patients with spinal muscular atropy.Ann Acad Med Singapore,2017,36(11):937-941.
[17]ELSHEIKH B,PRIOR T,ZHANG X,et al.Ananalysis of disease severity based on SMN2 copy number in adults with spinal muscular atrophy.Muscle Nerve,2009,40(4):652-656.
[18] 王旻晋,王 军,白梦鸽等,中国西南地区汉族脊肌萎缩症患者SMA相关基因分子特征,四川大学学报(医学版),2016,47(6):936-940。
[19] 美国FDA 政府公告,2016-12-23 夏训明编译.DB3C5DA0-053D-4195-A1E6-76518BF48BED
猜你喜欢
作文新天地(小学版)(2021年11期)2021-12-08
家庭百事通·健康一点通(2017年8期)2017-08-18
试题与研究·高考理综生物(2016年4期)2017-03-28
中外医学研究(2016年35期)2017-02-28
中国实用医药(2016年9期)2016-05-17
小溪流(画刊)(2016年6期)2016-05-14
百科知识(2015年18期)2015-09-10
小星星·阅读100分(高年级)(2015年4期)2015-05-26
小雪花·成长指南(2009年10期)2009-12-04
雕塑(1996年4期)1996-07-12
