
基于化学计量学的多花黄精多糖部分酸水解产物PMP-HPLC 指纹图谱构建
2022-07-08 13:54周忠瑜杜泽飞蒲婷婷杨丽英晏仁义段宝忠
食品工业科技 2022年13期
周忠瑜,杜泽飞,蒲婷婷,杨丽英,晏仁义,段宝忠,
(1.大理大学药学院,云南大理 671000;2.云南省农业科学院药用植物研究所,云南昆明 650205;3.湖北汉江大健康产业有限公司,湖北十堰 442521)
多花黄精为百合科黄精属植物Polygonatum cyrtonemaHua.,又名姜状黄精,为2020 年版《中华人民共和国药典》黄精药材的基原植物之一,是目前市场上黄精的主流栽培品种,具有补气养阴,健脾,润肺,益肾之功能[1],多花黄精为重要的药食同源资源,由于其良好的功效,近年来被广泛用于食品和保健品开发[2]。现代研究表明,多花黄精化学成分主要包括多糖,甾体皂苷、黄酮等成分[3−5],其中多糖是多花黄精中的质量标志物,也是中国药典黄精的质量控制指标[1],具有抗衰老、降血糖、降血脂、抗菌等药理作用[6−8]。据对保健食品原料的调查发现,黄精在中药类原料的使用频率排名靠前[9],目前市场上存在大量的黄精多糖产品如黄精膏、黄精饮料等产品,因此判定多花黄精产品的质量好坏和原料真伪极为重要。现有方法一般采用苯酚-硫酸法和蒽酮-硫酸法测定总多糖含量[10],用于控制多花黄精多糖产品质量,但此类方法选择性较差,不能有效地鉴别出掺假品;因此,迫切需要制定有效可靠的评价标准,以便科学地评价多花黄精多糖的质量,对黄精多糖保健食品的应用开发具有深远意义。
指纹图谱是近年来用于中药质量控制的一种有效、可行的方法,已广泛用于中药质量控制、药效成分研究领域[11−13]。由于黄精多糖为非单一分子化合物,结构复杂,且不含有共轭结构,使用常规高效液相二极管阵列检测法(High-performance liquid chromatography-diode array detection,HPLC-DAD)方法无法有效检测。已有学者采用高效液相色谱-蒸发光散射法、高效阴离子交换色谱-脉冲安培法和电喷雾式检测器(Charged aerosol detector,CAD)对植物多糖的单糖组成进行了研究[14−16],但蒸发光散射检测的耐用性和稳定性欠佳[17−18],脉冲安培检测的成本较高、稳定性差且对色谱条件要求较为严格[16],CAD 检测仪器价格昂贵,使用受到一定的限制[19−21]。有关黄精法定基原植物的指纹图谱研究方面,目前已有学者对滇黄精P.kingianumColl.Et Hemsl.和黄精P.sibiricumRed.多糖的指纹图谱进行了研究[22−23];而对于多花黄精P.cyrtonema的指纹图谱研究方面,仅见周宝珍,杨青等对多花黄精乙醇提取物的指纹图谱研究[24−25],尚未见多花黄精中的多糖组分的指纹图谱的报道。因此,建立能表征多花黄精中多糖指纹的方法具有重要意义。鉴于此,本文采用柱前衍生化-HPLC 对多花黄精药材的指纹图谱和单糖组成进行研究,并结合相似度分析(Similarity analysis,SA)、聚类分析(Hi-erarchical cluster analysis,HCA)和主成分分析(Prin-cipal component analysis,PCA)等化学计量学[26−28]手段,以期为多花黄精药材的质量控制奠定科学基础。
1 材料与方法
1.1 材料与仪器
黄精药材 13 批样品 购于安徽、四川、湖南等地,均为栽培品,经大理大学段宝忠教授鉴定为百合科植物多花黄精P.cyrtonemaHua.,新鲜根茎切片后于40 ℃烘箱烘干,样品信息详见表1,凭证标本保存于大理大学中药标本馆;葡萄糖(批号171106)、甘露糖(批号170921)、盐酸氨基葡萄糖(批号171210)、半乳糖(批号171206)、L-鼠李糖(批号171024)、L-岩藻糖(批号170813)、葡萄糖醛酸(批号170730)、木糖(批号170912)、半乳糖醛酸(批号170903)、核糖(批号171103)、阿拉伯糖(批号171219) 以上单糖纯度≥98%,购自上海融禾医药科技有限公司;1-苯基-3-甲基-5-吡唑啉酮(1-pheny-3-methyl-5-pyrazolone,PMP) 阿拉丁;色谱级乙腈(批号085884) Fisher Scientific;水为超纯水,其他试剂为国产分析纯。
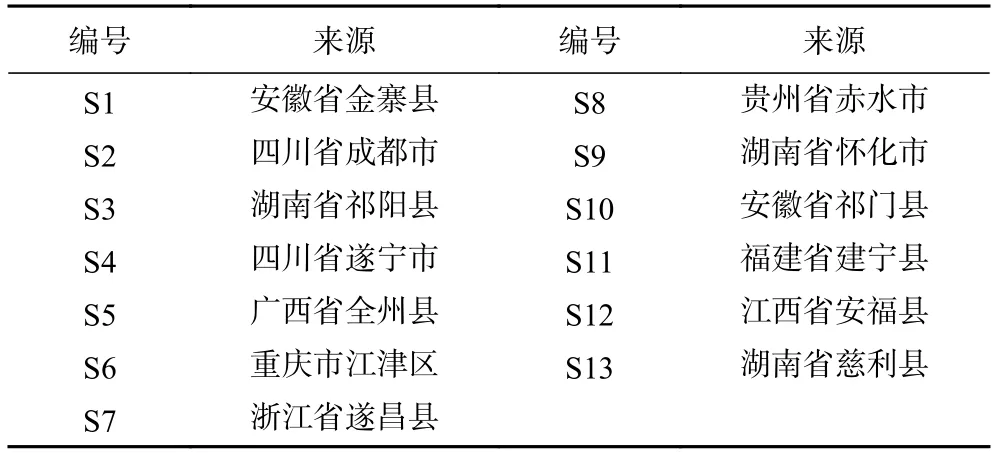
表1 样品信息Table 1 Information of samples
Agilent 1200 高效液相色谱仪 美国安捷伦科技有限公司;GH-252 电子天平 日本AND 公司;AL204 电子天平 梅特勒-托利多仪器(上海)有限公司;SB25-12D 超声波清洗机 宁波新芝生物科技股份有限公司。
1.2 实验方法
1.2.1 色谱条件 Agilent Zorbax SB-C18色谱柱(250 mm×4.6 mm,5 μm);流动相:乙腈(A)−0.025 mol·L−1磷酸盐缓冲溶液(B)(pH7.5);梯度洗脱(0~10 min,15%~17% A;10~18.5 min,17%~22.5%A;18.5~20 min,22.5%~23.5% A;20~32 min,23.5%~30% A);流速:0.8 mL·min−1;柱温:35 ℃;检测波长:250 nm;进样量:20 μL。
1.2.2 衍生化对照品溶液制备 精密称取各单糖对照品适量,用蒸馏水配制成浓度约为0.5 mg·mL−1的单糖对照品溶液。精密吸取上述对照品溶液400 μL,置于5 mL 的安瓿瓶中,精密加入200 μL 0.6 mol·L−1NaOH 和0.5 mol·L−1PMP 溶液,置70 ℃条件下反应60 min。取出,放冷,精密加入200 μL 0.6 mol·L−1HCl 溶液,混匀。加入等体积的三氯甲烷,混匀,离心10 min(4000 r·min−1),去掉三氯甲烷层,重复多次至三氯甲烷层无色,即得衍生化对照品溶液。
1.2.3 供试品溶液制备
1.2.3.1 粗多糖制备 取多花黄精样品于40 ℃烘箱干燥12 h 至恒重,粉碎,过80 目筛,取粉末5.0 g,精密称定,根据预实验得到最优工艺制备粗多糖,即加入100 mL 水于80 ℃下超声提取,重复3 次,每次1 h,抽滤,洗净滤渣,浓缩至10 mL,放入离心机离心20 min(4000 r·min−1)。取上清液转移至分液漏斗,加入3 倍体积的石油醚进行萃取,直至石油醚层无色,静置分层,取下层水相,调节pH 至6,加入体积分数为2%的木瓜蛋白酶溶液(80 万U/g),水浴温度60 ℃,酶解4 h,待反应完成,沸水浴灭酶10 min。取上清液采用Sevage 法(正丁醇:氯仿=1:5)脱去蛋白,直至无絮状物生成,离心10 min(4000 r·min−1),再取上清液转至烧杯中,精密缓慢的加入6 倍量的无水乙醇,快速搅拌,于4 ℃冰箱中放置12 h,离心,向沉淀中加10 mL 95%乙醇,洗涤2 次,再离心,加热水使沉淀溶解,转移至10 mL 量瓶中,室温下放冷,后定容,再将溶液置于烧杯中冷冻干燥,最后得粗多糖粉末。
1.2.3.2 衍生化供试品溶液制备 精密称取5 mg 粗多糖粉末,置于5 mL 的安瓿瓶中,精密加入2 mL 4 mol·L−1三氟乙酸(TFA)溶液,封口,置于110 ℃条件下7 h,进行水解,取出,室温放冷,后水浴蒸干,向残渣中加入甲醇1 mL,烘干,重复多次,直至三氟乙酸除尽。再加热水适量,使沉淀溶解,转移至1 mL容量瓶中,室温放冷,定容摇匀,得供试品酸水解溶液。取酸水解溶液400 μL,按1.2.2 项下衍生化方法制备,得衍生化供试品溶液,每批样品重复3 次。
1.2.4 总多糖含量测定 按课题组已发表文献方法,对13 批多花黄精总多糖含量进行测定[10]。
1.3 数据处理
相似度分析采用《中药色谱指纹图谱相似度评价系统》(2.0 版),聚类分析采用SPSS 20.0 软件进行,主成分分析采用SIMCA 13.0 软件进行。
2 结果与分析
2.1 色谱条件的选择
本研究比较了以下4 种色谱柱:Zorbax SB-C18(4.6 mm×250 mm,5 μm),Zorbax Extend-C18(4.6 mm×250 mm,5 μm),Zorbax SB-Aq-C18(4.6 mm×250 mm,5 μm),Zorbax XDB-C18(4.6 mm×250 mm,5 μm)。实验结果表明,Agilent Zorbax SB-C18色谱柱(4.6 mm×250 mm,5 μm)出峰较多,各成分分离较好。同时,当流动相为纯水时,部分色谱峰产生了拖尾情况,因此,在实验中考察不同浓度磷酸盐缓冲溶液(0.01、0.02、0.025 mol·L−1)对色谱峰分离效果的影响,结果显示,当磷酸盐缓冲溶液浓度为0.025 mol·L−1时,所得色谱峰峰形及分离度较好,故选择0.025 mol·L−1的磷酸盐缓冲溶液作为洗脱溶剂。
2.2 方法学考察
2.2.1 精密度试验 取同一批供试品溶液(S2),按1.2.1 项下方法连续进样6 次,测得各共有峰相对保留时间和相对峰面积的RSD 均小于2.10%,表明精密度良好。
2.2.2 重复性试验 取同一批S2 样品6 份,按1.2.3项下方法进行制备,在1.2.1 项色谱条件下进样分析。测得结果显示,各共有峰的相对保留时间与相对峰面积的RSD 均小于2.60%,表明重复性良好。
2.2.3 稳定性试验 取同一批S2 供试品溶液,分别在0、4、8、12、18、24、48 h 进样测定,测得结果显示,各共有峰的相对保留时间与相对峰面积的RSD均小于2.70%,表明供试品溶液在48 h 内稳定。
2.3 指纹图谱的构建
将13 批多花黄精样品按1.2.3 方法制备供试品溶液,按1.2.1 项色谱条件下检测,记录色谱图,色谱数据导入《中药色谱指纹图谱相似度评价系统2.0 版》软件,采用中位数法,以S1 批样品作为参照谱进行指纹匹配,生成共有模式指纹图谱(图1),可见共有10 个共有峰。相似度结果见表2,13 批多花黄精的指纹图谱相似度在0.781~0.945 之间,其中S1(安徽省金寨县)、S3(湖南省祁阳县)、S8(贵州省赤水市)、S11(福建省建宁县)和S12(江西省安福县)5 批样本相似度大于0.90,其它样品低于0.90,占61.5%,这一结果与黄精P.sibiricumRed.和滇黄精P.kingianumColl.et Hemsl.的HPLC 指纹图谱研究结果不一致[22−23],其原因是否是产地或生长年限等影响了其多糖组成,还有待进一步深入研究。本研究中采集自湖南省慈利县的S13 号相似度最低为0.781,观察发现与其他样品相比,S13 号样品根茎较幼嫩,已有研究表明黄精幼嫩部位的多糖含量较成熟部位低[29],这可能是其相似度较低的原因。
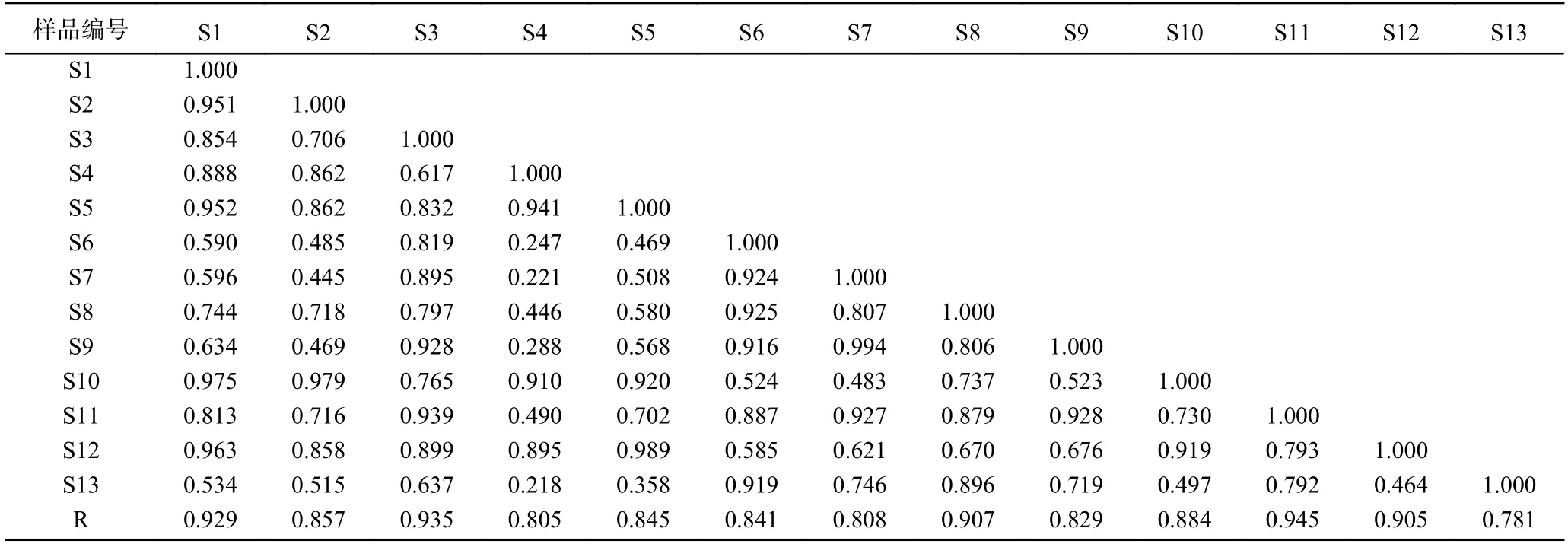
表2 13 批不同产地多花黄精相似度Table 2 Similarity evaluation of 13 batches of P.cyrtonema from different localities
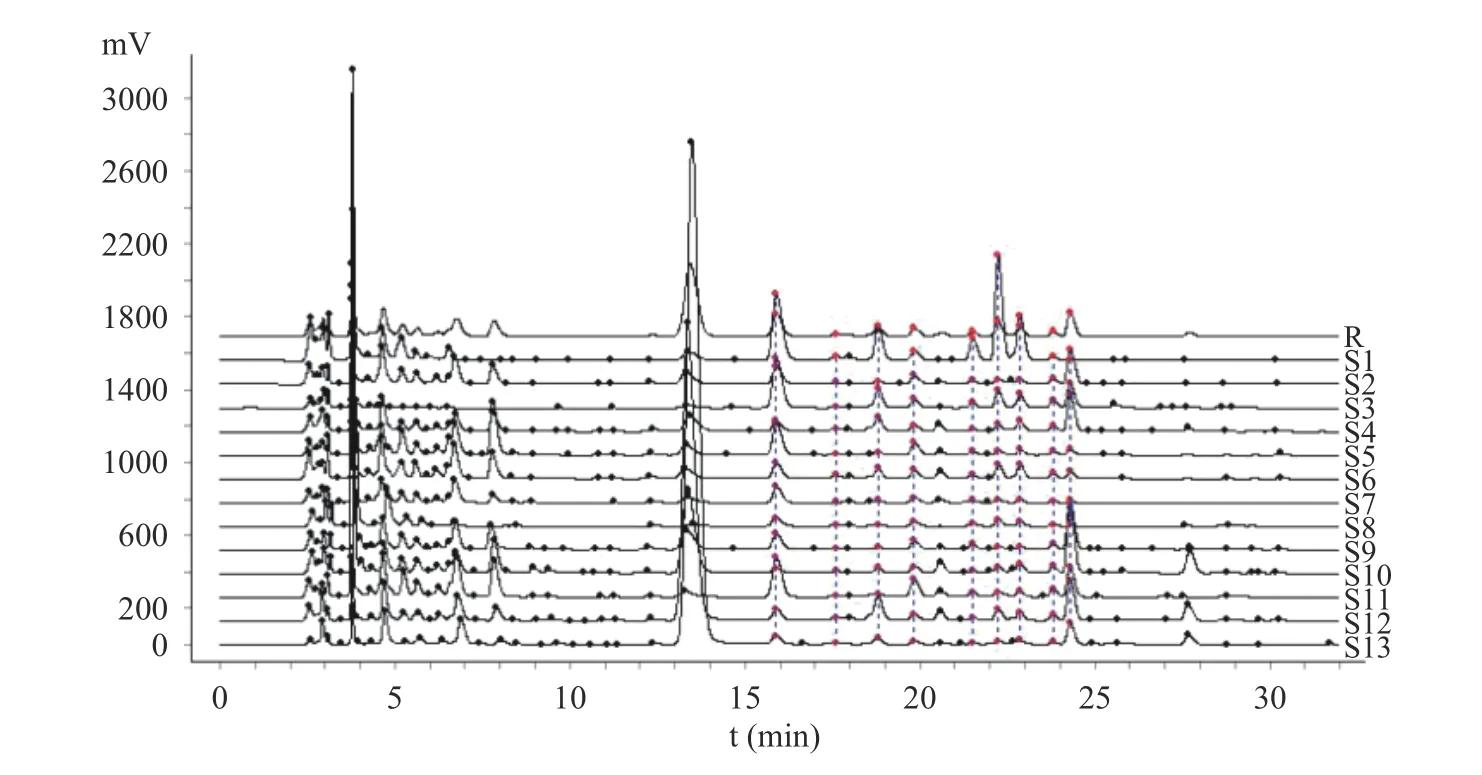
图1 13 批多花黄精多糖柱前衍生化HPLC 指纹谱Fig.1 Pre-column derivation HPLC characteristic fingerprint of polysaccharide hydrolysate from 13 batches of P.cyrtonema
2.4 单糖定性鉴定
样品及单糖对照品的PMP-HPLC 色谱图见图2。通过与对照品的保留时间比对,发现10 个共有峰中,5 个与单糖对照品保留时间一致,即4 号峰(18.80 min)为半乳糖醛酸,6 号峰(20.13 min)为葡萄糖醛酸,7 号峰(21.50 min)为半乳糖,8 号峰(22.22 min)为葡萄糖,9 号峰(22.84 min)为木糖,单糖研究结果与王坤等[30]对多花黄精单糖组成研究结果基本一致。在前期研究中,何连军等[14]采用高效阴离子交换色谱-脉冲安培检测法,检测到多花黄精含有果糖,而本研究未检测到果糖,主要原因是由于PMP 仅能与醛糖发生衍生化反应,果糖属于酮糖,不发生衍生反应[31]。
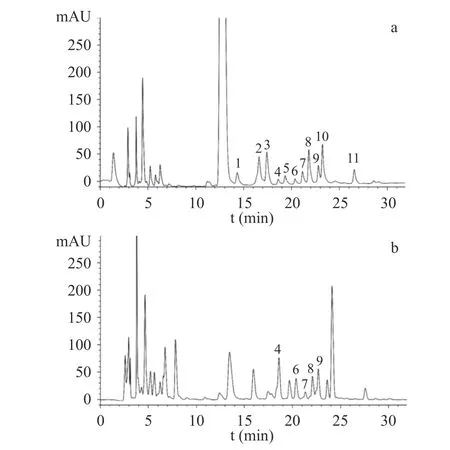
图2 多花黄精药材共有模式图谱及对照品HPLC 图Fig.2 HPLC fingerprints of P.cyrtonema and chromatograms of reference substances
2.5 总多糖含量
多糖是黄精药材的重要质量标志物[32],按2020年版《中华人民共和国药典》含量测定项规定,其干燥品含多糖以无水葡萄糖(C6H12O6)计,不得少于7.0%。本研究显示,13 批多花黄精药材的多糖含量符合要求,在7.18%~16.27%范围内,结果见表3。可见总多糖含量最高的S3 号(湖南省祁阳县),最低是S9 号(湖南省怀化市),约相差1 倍,表明不同产地多花黄精多糖含量有明显差异,与相似度分析结果一致。已有研究表明,产地和生长年限是影响黄精多糖含量的重要影响因素[29,33],本研究中,样品为随机采集,产地和生长年限可能是形成其多糖差异的原因。
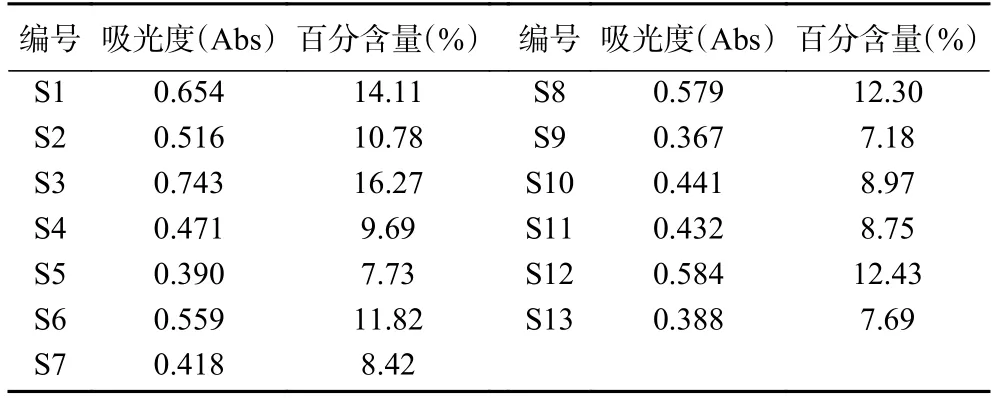
表3 13 批多花黄精的总多糖含量测定结果Table 3 Determination results of total polysaccharide content of 13 batches of P.cyrtonema
2.6 聚类分析
聚类分析(HCA)是按照个体数据特征对样本进行分类的一种方法,其同一类别个体间具有较高的相似度[11]。将13 批样品单位质量药材峰面积进行量化,得到13×10 阶的数据矩阵,采用SPSS 20.0 版软件,瓦尔德法(Ward)进行分析,结果见图3。可见当判别条件距离为10 时,13 批多花黄精药材被分成两类,I 类包括12 个样品,为S1~S12 号,II 类仅有S13 号(湖南省慈利县),若以相似度0.80 为界限,聚类结果与相似度和多糖含量结果一致。不同产地的样品在聚类图上无明显区分,表明栽培多花黄精药材的多糖成分类别差异不大,S13 号样本偏离的原因可能是采收年限、种质资源或栽培技术差异造成的[34]。
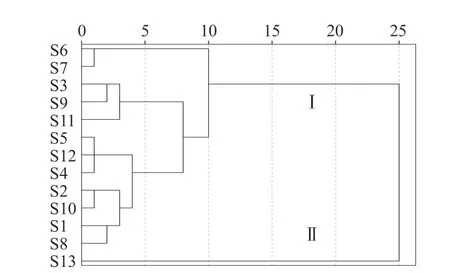
图3 不同产地多花黄精系统聚类分析Fig.3 The cluster analysis of P.cyrtonema from different localities
2.7 主成分分析
主成分分析(PCA)是将多维具有相关性的数据压缩为少数几个相互独立数据的统计方法,其在不损失主要信息的前提下实现降维,扩大样本之间的差异,可解决由于中成药成分复杂所致的普带重叠分析困难[35]。为了更加全面、系统地分析13 批多花黄精之间的差异,揭示其内在关系,将上述13×10 数据矩阵采用SPSS 20.0 软件计算特征值和方差贡献率,见表4,以主成分的特征值大于1 和累计方差贡献率大于85%,作为选择主成分因子依据[36],结果显示前三个主成分可代表89.364%的信息量。采用SIMCA-P13.0 计算PCA 得分图,结果见图4,13 个样品被分成两类,其中采集于湖南省慈利县的S13 号样品单独聚为一类,与HCA 结果一致,观察发现与其他样品相比,S13 号样本较为幼嫩,可能是其单独聚为一支的原因。
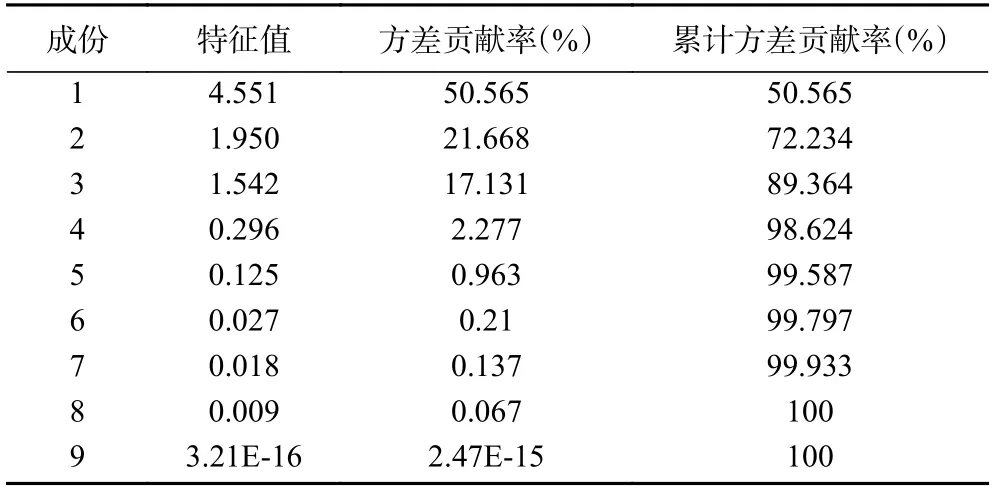
表4 特征值和方差贡献率Table 4 Characteristic value and variance contribution rate
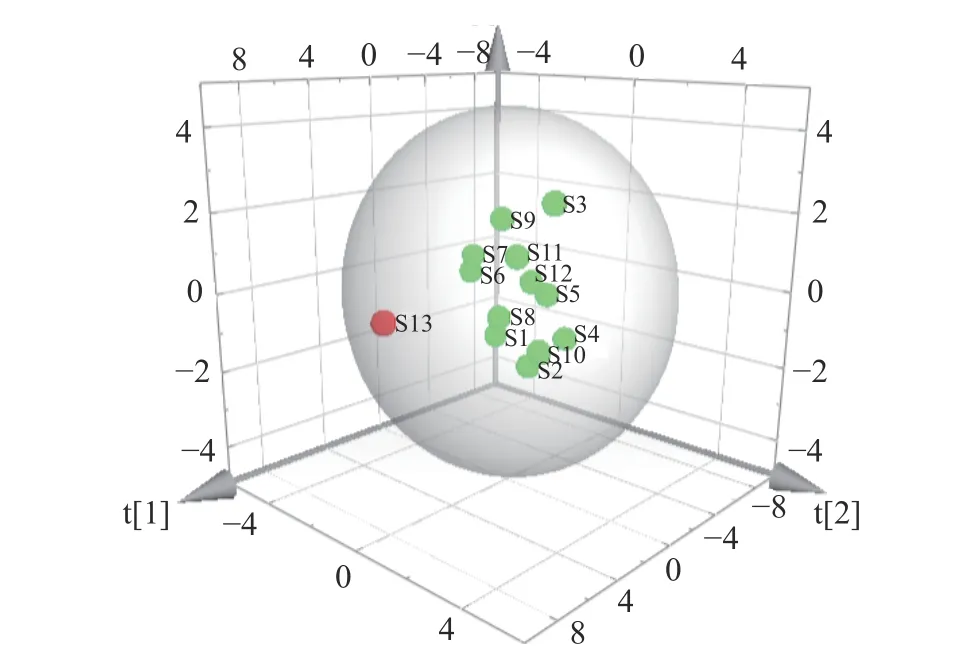
图4 多花黄精PMP-HPLC 图谱PCA 结果Fig.4 PCA results of PMP-HPLC fingerprints of P.cyrtonema
3 结论
本实验首次建立了不同产地多花黄精中多糖成分的PMP-HPLC 指纹图谱,并对其单糖组成进行了研究,在此基础上,结合指纹图谱技术,采用相似度分析,聚类分析和主成分分析对其指纹图谱进行了研究,并对其总多糖含量进行了测定。实验结果表明,13 批多花黄精单糖组成均含半乳糖醛酸、葡萄糖醛酸、半乳糖、葡萄糖、木糖,其总多糖含量范围在7.18%~16.27%之间,HCA 和PCA 分析结果一致,样本被分为2 类,其次,13 批多花黄精的指纹图谱相似度较低,在0.781~0.945 之间,其中相似度低于0.90 的样品占所研究样品的61.5%,从一定程度反映了多花黄精药材质量的不均一,这一结果与黄精其他基原物种研究结果不一致[22−23],其原因可能是种质资源、产地或生长年限等影响了其多糖生成,还有待进一步深入研究。综合产地与指纹图谱相似度、总多糖含量、单糖组成等分析,未发现明显的规律,已有研究表明,多花黄精不同龄节间的有效成分含量差异较大[37],不同生长年限的黄精药材其多糖成分亦存在一定差异[29],提示采收年限和栽培技术可能是造成多花黄精品质差异的原因。因此,为确保黄精药材临床用药的有效和安全,有必要建立多花黄精的规范化栽培技术体系,以确保多花黄精药材品质的一致性。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
中老年保健(2021年11期)2021-08-22
Digital Chinese Medicine(2020年3期)2020-11-03
云南农业科技(2020年5期)2020-10-14
中成药(2019年12期)2020-01-04
中国科技纵横(2018年2期)2018-11-29
中国中药杂志(2017年20期)2017-11-11
科技资讯(2017年20期)2017-08-22
分析化学(2015年10期)2015-11-03
中国医药导报(2011年27期)2011-12-31
