
克白合剂HPLC 指纹图谱及有害残留物检测*
2022-07-15 02:17童菊华黄巧玲吴经耀
药学与临床研究 2022年3期
童菊华,黄巧玲,王 迅,吴经耀,徐 强
1 浙江省中药研究所有限公司,杭州 310023;2 杭州市第三人民医院 药剂科,杭州310009
克白合剂是杭州市第三人民医院院内制剂,为临床治疗白癜风多年的经验方,由补骨脂、菟丝子、白芷等组成,对于气滞血瘀、肝肾阴虚型白癜风具有确切的疗效[1]。目前该制剂的执行标准为《浙江省医疗机构制剂规范(2005 年版)》,包括性状、何首乌与枸杞子薄层鉴别、相对密度、pH 值、合剂项下等常规的定性检验项目,缺少定量控制指标以及制剂整体质量的一致性评价技术。
克白合剂为16 味中药组成的大复方,所含的化学成分十分复杂,仅用指标成分的含量测定控制其质量,难以评价制剂生产过程中的整体质量情况[2]。而中药指纹图谱技术是评价中药优劣、鉴别真伪和确保其一致性和稳定性的有效方法。
现代分析技术的飞速发展,中药指纹图谱技术在研究中药有效成分、控制中药质量以及鉴别方面也越来越成熟,可促进中药走向现代化[3]。因此,在增加了补骨脂素与异补骨脂素含量测定指标的基础上[4,5]进一步对克白合剂指纹图谱进行了研究,并对其外源性有害物质进行了控制研究[6,7],为复方制剂的全面质量控制探索一条可行之路,为其临床用药的安全性与有效性提供质量保障。
1 仪器与药品、试剂
Agilent 1100 高效液相色谱系统,Agilent 6980N 气相色谱仪(均美国安捷伦公司);Mars 6 微波消解仪(美国CEM 公司);AA-7000 原子吸收分光光度计(日本岛津公司);高压蒸汽灭菌器(上海施都凯公司);生化培养箱、霉菌培养箱(均上海精宏实验设备公司);ME204-E 电子分析天平(瑞士梅特勒公司);KQ-500VDE 型三频数控超声波清洗器(昆山市超声仪器公司);旋转蒸发仪(海亚荣生化仪器厂)。
补骨脂素对照品、异补骨脂素对照品(补骨脂素批号:110739-201617,纯度,99.7%;异补骨脂素批号:110738-201715,纯度,99.5%;均中国食品药品检定研究院);金丝桃苷对照品(批号111521-201708,纯度95.1%),特女贞苷对照品(批号11926-201605,纯度93.3%),均中国食品药品检定院。
甲醇、乙腈为色谱纯;甲醇为分析纯,其余试剂均为分析纯;水为超纯水。
2 方法与结果
2.1 克白合剂对照指纹图谱方法的建立
2.1.1 色谱条件与系统适用性试验 色谱柱:ZORBAX SB C18柱(250 mm × 4.6 mm,5 μm,Agilent Technologies);流动相:乙腈为流动相A,0.2%磷酸溶液(v∶v)为流动相B,按表1 进行梯度洗脱;柱温:30 ℃;检测波长:246 nm;流速:1 mL·min-1。
理论板数按补骨脂素峰计算应不低于3000;补骨脂素峰和异补骨脂素峰分离度均应>1.5。
2.1.2 对照品溶液配制 取补骨脂素对照品适量,精密称定,加甲醇制成每毫升含0.15 mg 的溶液,即得。
另取异补骨脂素对照品、金丝桃苷对照品、特女贞苷对照品适量,精密称定,分别加甲醇制成每毫升各含金丝桃苷0.2 mg、特女贞苷0.25 mg、异补骨脂素0.1 mg 的溶液,即得,用于指纹图谱中各色谱峰化学成分的确认。
2.1.3 供试品溶液制备 精密量取克白合剂10 mL,加入流动相稀释,定容至25 mL 量瓶中,摇匀,离心,滤过,取续滤液,即得。
2.1.4 专属性试验 按照“2.1.1”项下的色谱条件,精密吸取混合对照品溶液与供试品溶液各10 μL,注入液相色谱仪,得到相应的色谱图。
区间左线施工完成后,左侧0#承台的1号、2号角点X向位移最大约为0.82 mm,3号、4号角点X向位移最大约为0.78 mm。右侧1#承台的1号、2号角点X向位移最大约为0.54 mm,3号、4号角点X向位移最大约为0.50 mm。
供试品指纹图谱中,在与对照品色谱图相对应的保留时间处有色谱峰出现。见图1、图2。

图1 混合对照品高效液相色谱图
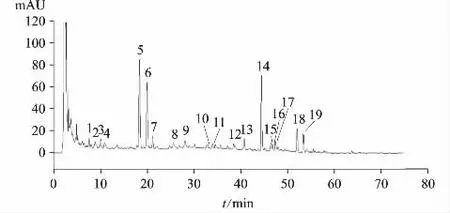
图2 克白合剂供试品溶液指纹图谱(批号180627)
2.1.5 指纹图谱相似度评价方法 将色谱图数据以AIA(即*.cdf)的格式文件导入《中药色谱指纹图谱相似度评价系统》2.0 版进行评价,以S1 为参照图谱(批号180627),采用中位数法,时间窗宽度为0.1,进行全谱峰匹配,系统根据10 批次样品色谱图的共有模式生成克白合剂对照指纹图谱R,计算指纹图谱的相似度。
2.2 方法学考察
2.2.1 精密度试验 取克白合剂样品适量(批号180627),按照“2.1.3”项下的方法制备供试品溶液,重复进样6 次。以18 号色谱峰为参照,经计算得到各共有峰的相对保留时间的RSD 均<0.32%,各相对峰面积的RSD 均<2.78%,表明仪器精密度良好。
2.2.2 重复性试验 取克白合剂样品适量(批号180627),按照“2.1.3”项下的方法,平行制备6 份供试品溶液,分别进样测定。以18 号色谱峰为参照,经计算得到各共有峰的相对保留时间的RSD 均<0.37%,各相对峰面积的RSD 均<2.84%,表明方法重复性良好。
2.2.3 稳定性试验 取同一供试品溶液适量(批号180627),室温放置,分别于0、3、6、9、12、24 h 进样。以18 号色谱峰为参照,经计算得到各共有峰的相对保留时间的RSD 均<1.12%,各相对峰面积的RSD均<2.91%,表明供试品溶液在24 h 内稳定性良好。
2.3 指纹图谱检测及共有峰归属
将10 批克白合剂样品按“2.1.3”项下方法制备供试品溶液,并按“2.1.5”项下相似度评价方法,生成对照指纹图谱R,见图3,计算相似度。各批次样品的指纹图谱全谱图与对照图谱R 相比,相似度结果见表2。结果10 批克白合剂的指纹图谱相似度均>0.9,整体相似性较好,说明克白合剂的制备工艺稳定,整体质量差异不大。
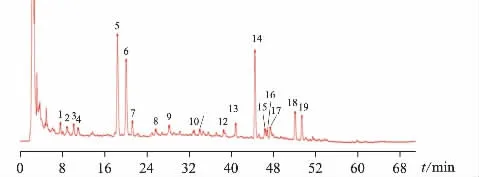
图3 克白合剂对照指纹图谱
根据共有峰的归属,在不同批次的克白合剂指纹图谱中,女贞子、丹参、补骨脂、白芷、黄芪、独活对共有峰的贡献较大;蜈蚣、地黄、刺蒺藜、党参、枸杞子及地龙的特征峰未能体现,可能与合剂中水溶性成分较多,而极性小的的成分在制备过程中转移率较低有关。
3 克白合剂微生物限度内源性及外源性有害残留物质的安全性研究限量检查
3.1 微生物限度
取不同批次的克白合剂样品10 批,按《中国药典》2020 年版四部通则1105~1107 检查,结果见表2。表明10 批克白合剂的微生物限度测定结果符合现执行标准。
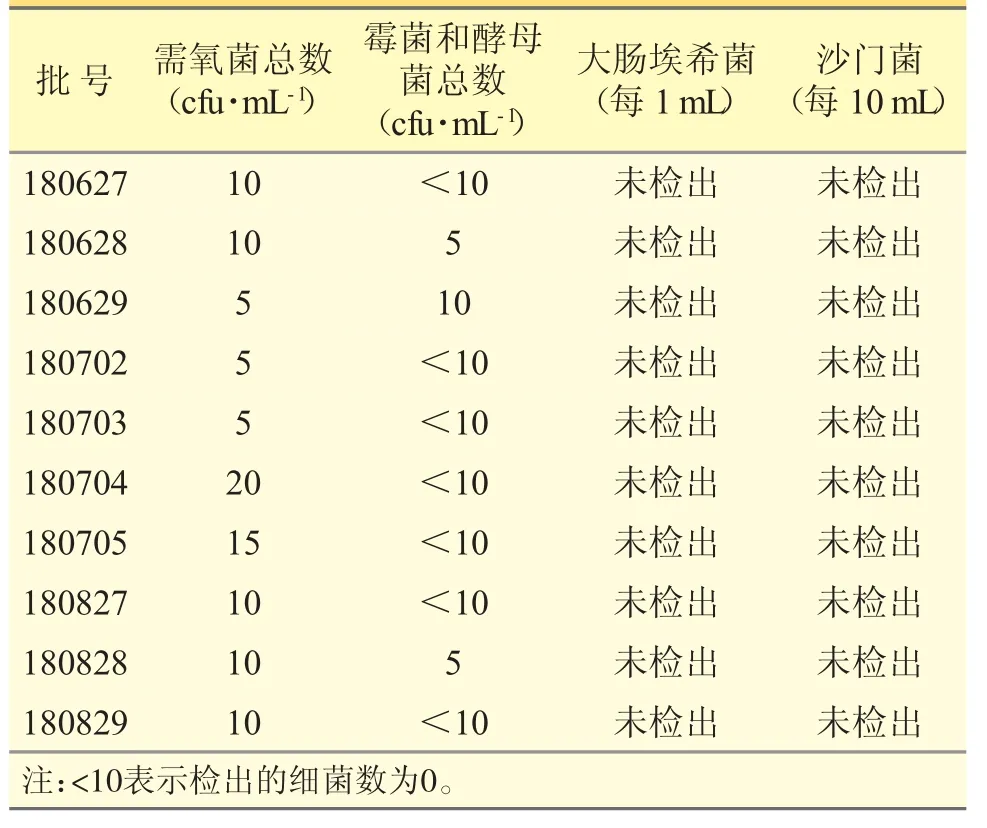
表2 克白合剂微生物限度测定结果
3.2 重金属及有害元素测定
取不同批次的克白合剂样品10 批,按《中国药典》2020 年版四部通则2321 原子吸收分光光度法对铅、镉、砷、汞、铜进行测定,结果见表3。
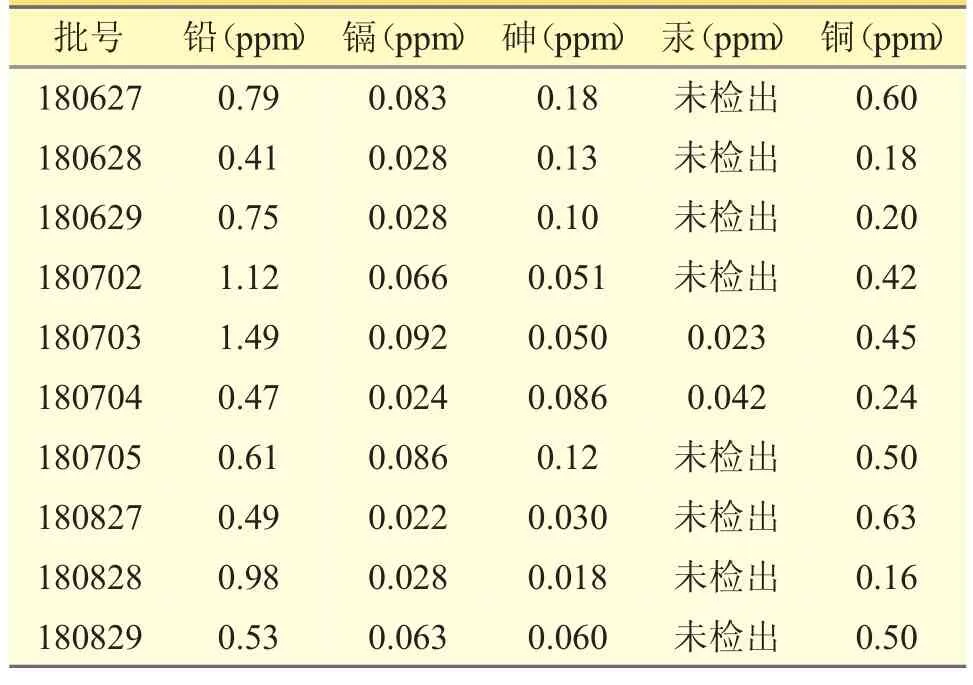
表3 克白合剂重金属及有害元素测定结果
10 批次克白合剂的重金属及有害元素含量较低,均符合2020 年版《中国药典》规定中相关的限度执行标准。
3.3 有机氯类农药残留量测定
3.3.1 方法 按照《中国药典》2020 年版四部通则2341 第一法测定9 种有机氯类农药残留量。
3.3.1.1 对照品溶液的的制备 分别精密吸取六六六(α-BHC,β-BHC,γ-BHC,δ-BHC)、滴滴涕(PP'-DDE,PP'-DDD,OP'-DDT,PP'-DDT)及五氯硝基苯(PCNB)1mL,分别置于10mL 量瓶中,加石油醚稀释至刻度,摇匀,制得每毫升各含5000、5000、10000μg 的对照品储备液。精密量取上述各对照品储备液1mL,置25mL 量瓶中,用石油醚(60~90℃)稀释至刻度,摇匀,制得混合对照品储备液。再精密量取上述混合对照品储备液,用石油醚(60~90℃)制成每毫升分别约含0.39、0.78、1.56、3.12、6.25、12.50、25.00、50.00、100.00、200.00 μg 的混合对照品溶液。
3.3.1.2 供试品溶液的的制备 精密量取克白合剂10 mL,置100 mL 具塞锥形瓶中,加水10 mL 浸泡过夜,精密加丙酮40mL,称定重量,超声处理30min,放冷,再称定重量,用丙酮补足减失的重量,再加氯化钠6 g、二氯甲烷30 mL,称定重量,超声处理15 min,称定重量,用二氯甲烷补足减失的重量,静置(使分层),将有机相迅速移入装有适量无水氯化钠的100mL 具塞锥形瓶中,放置4 h。精密量取35 mL,于40 ℃水浴上减压浓缩至近干,加少量石油醚(60~90 ℃),如前反复操作至二氯甲烷及丙酮除净,用石油醚(60~90 ℃)溶解并转移至10 mL 具塞刻度离心管中,加石油醚(60~90 ℃)精密稀释至5 mL。小心加入硫酸1 mL,振摇1 min,3000 r·min-1离心10 min。吸取上清液即为供试品溶液。
3.3.2 色谱条件与系统适用性试验
3.3.2.1 专属性试验 按照 《中国药典》2020 年版四部通则2341 第一法“9 种有机氯类农药残留量测定法”项下色谱条件,精密吸取供试品溶液2 μL、对照品溶液1 μL,注入气相色谱仪,得到相应的气相色谱图。见图4、图5。
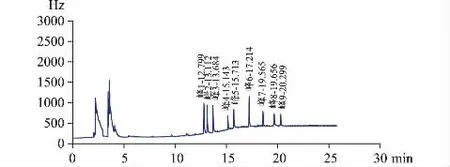
图4 9 种有机氯农药对照品气相色谱图
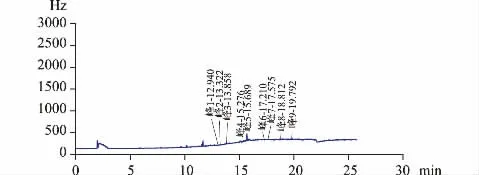
图5 样品气相色谱图
3.3.2.2 线性关系考察 精密吸取“3.3.1.1”项下各混合对照品溶液,每一浓度标样进样1μL,记录色谱图。以进样量(X)为横坐标,峰面积(Y)为纵坐标,进行线性回归,计算得回归方程如下:α-BHC(Y=245.8845X+2.1741,r=0.9999);β-BHC(Y=90.4858X+170.4813,r=0.9996);γ-BHC(Y=214.8878X+193.7824,r=0.9999);δ-BHC(Y=143.188 2X+169.618 7,r=0.999 9);PP'-DDE(Y=210.730 6X-34.786 2,r=1.000 0);PP'-DDD(Y=136.801 0X+105.891 3,r=0.999 9);OP'-DDT(Y=123.398 1X-69.741 8,r=0.999 9);PP'-DDT(Y=131.900 3X-228.276 0,r=0.999 9);α-PCNB(Y=190.888 2X+803.363 2,r=0.998 8)。
3.3.2.3 检测限 取低浓度混合对照品溶液,逐级稀释,进样测定,以S/N=3 时注入仪器的各对照品溶液的量确定检测限。检测限结果如下:α-BHC 为0.12μg·L-1,β-BHC 为0.21μg·L-1,γ-BHC 为0.12μg·L-1,δ-BHC 为0.13 μg·L-1;PP'-DDE 为0.13 μg·L-1,PP'-DDD 为0.23 μg·L-1,OP'-DDT 为0.98 μg·L-1,PP'-DDT 为1.30 μg·L-1;PCNB(五氯 硝基苯)为0.07 μg·L-1。
3.3.3 样品测定 分别精密吸取对照品溶液1 μL、供试品溶液2 μL,注入气相色谱仪,按对照品比较外标法计算供试品中9 种有机氯类农药残留量。结果见表4。
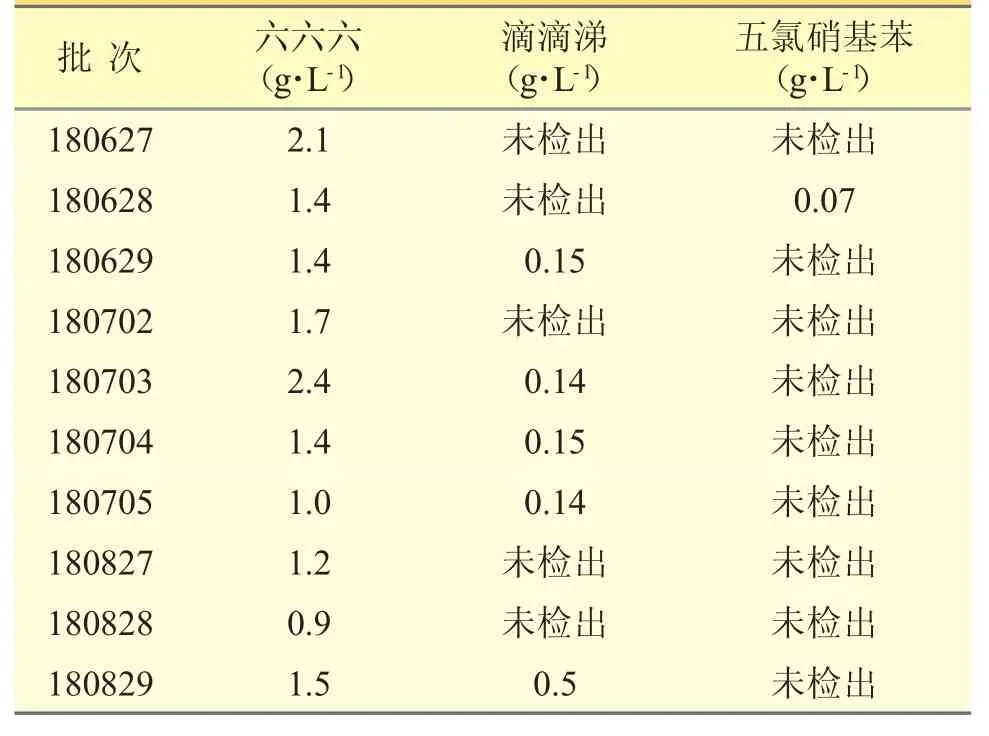
表4 克白合剂9 种有机氯类农药残留量测定结果
4 结论
4.1 指纹图谱色谱条件的选择
查阅有关复方制剂指纹图谱的研究文献[8],对克白合剂指纹图谱进行了研究。本课题经过一系列的色谱条件研究,发现流动相中的有机相比例达到55%后,图谱中基本无色谱峰出现,故现确定的色谱条件能对主要色谱峰达到较好分离,且便于进行制剂-药材的谱峰归属分析,能较大限度地反映其特征。
4.2 色谱峰归属性分析
在不同批次的克白合剂指纹图谱中,女贞子、丹参、补骨脂、白芷、黄芪、独活对共有峰的贡献较大;蜈蚣、地黄、刺蒺藜、党参、枸杞子及地龙的特征峰未能体现,这可能与制备过程中各成分转移率不同有关。研究建立的指纹图谱鉴别方法简便、重复性好,能达到控制批间质量一致性的目的,并为克白合剂的综合质量评价提供了科学依据。
4.3 微生物、重金属有害元素及农药残留量的限量测定
在对制剂中有效性成分进行质量控制的同时,关注影响制剂使用安全的外源性有害成分研究。对克白合剂中的微生物、重金属及有害元素、农药残留量进行测定,其中对重金属及有害元素进行了检测限的确定,均符合药典要求,可保证克白合剂在临床长期使用中更为安全。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
草业科学(2022年9期)2022-10-21
医学概论(2022年3期)2022-04-24
河南农业·综合版(2022年2期)2022-03-18
河南农业(2022年2期)2022-03-14
河南农业·综合版(2021年7期)2021-08-23
果农之友(2014年2期)2014-10-21
中国医药导报(2011年27期)2011-12-31
农家科技(2011年3期)2011-06-13
农村百事通(2009年6期)2009-05-13
