
不同酶切方式引发大豆蛋白构象变化及功能特性评价
2022-07-23 10:16郑环宇赵晓明孙远达和铭钰
中国粮油学报 2022年6期
郑环宇, 赵晓明, 张 梦, 孙远达, 和铭钰, 李 杨, 滕 飞
(东北农业大学食品学院1,哈尔滨 150030) (黑龙江省绿色食品科学研究院2,哈尔滨 150028) (国家大豆工程技术研究中心3,哈尔滨 150000)
大豆是世界重要粮食作物之一,因其高蛋白、低脂肪含量及低生产成本成为优质蛋白来源[1]。大豆蛋白营养价值高,持水性、凝胶性等加工特性应用广泛,但大豆蛋白易受环境影响,溶解性及抗氧化性能较差[2],限制其作为低成本、高品质乳化剂在食品加工中的应用。酶促水解是改善大豆蛋白利用率的有效途径,其通过酶催化作用将蛋白分解成小分子物质,作用条件温和,保护蛋白营养价值,有效降低大豆蛋白致敏性,提高功能特性,是一种极具潜力的蛋白改性方法。
蛋白酶法改性过程受酶水解时间、pH、温度及酶的专一性等因素影响,其中酶的专一性是影响肽的数目及相应的酶切位点的关键因素之一,不同酶水解获得蛋白水解物的乳化性及其抗氧化性对蛋白乳化剂在加工中的影响不可忽视。依据蛋白酶活性中心关键性氨基酸可将其分为4类,即丝氨酸蛋白酶、半胱氨酸蛋白酶、金属蛋白酶及天门冬氨酸蛋白酶[3]。枯草杆菌碱性蛋白酶及胰蛋白酶均为常见丝氨酸蛋白酶,其活性中心处丝氨酸内部的亲核基团常与底物亲电基团发生共价催化,产物功能特性得到显著改善。大豆蛋白经枯草杆菌丝氨酸蛋白酶水解后,其乳化性、起泡性及表面疏水特性均有改善[4]。大豆蛋白经胰蛋白酶进行部分肽键断裂,结构复杂的蛋白质分子展开成多肽链,暴露埋藏于蛋白内部的具有抗氧化性氨基酸残基,发挥其清除自由基能力[2]。胰蛋白酶与胃蛋白酶及碱性蛋白酶2.4 L FG水解大豆蛋白获得的水解产物相比具有最高的ABTS+清除活性,且与水解时间不相关[5]。菠萝蛋白酶的活性中心为半胱氨酸,其通过内部富含电子的基团攻击底物亲电子基团,进而发挥共价催化作用。经菠萝蛋白酶处理黑豆蛋白水解产物能显著抑制亚油酸体系中过氧化物形成,增强水包油乳液氧化稳定性,可以作为食品中天然抗氧化剂成分[6]。金属蛋白酶类常以酶原形式出现,其活性中心处保留定量金属离子,既可以维持酶活性,又参与金属催化反应,促进底物在反应中正确定向。枯草杆菌中性蛋白酶作为典型的金属蛋白酶,其断裂由疏水性大分子的氨基酸的羧基形成的肽键[3],将大分子蛋白分解为小分子肽段,改善溶解度,降低黏度,提升性能及生物效价[4]。胃蛋白酶为活性中心含天门冬氨酸的酸性内切蛋白酶,内部两个天门冬氨酸残基交替充当酸碱催化剂,进行酸碱催化反应。经胃蛋白酶水解制得大豆蛋白水解产物中β亚基相对组成最高,显示出良好的混合乳液稳定性和快速融化速率[7]。酶的专一性是影响产物功能特性的重要因素,但有关酶的专一性对大豆蛋白功能特性的影响及其与构象变化相关性的研究还有待探索。
本实验以5种商用酶制剂(枯草杆菌碱性蛋白酶、菠萝蛋白酶、枯草杆菌中性蛋白酶、胃蛋白酶和胰蛋白酶)水解大豆蛋白产物为研究对象,对水解产物的理化指标、二级结构、亚基组成进行分析,同时对其乳化性、抗氧化性进行评价,旨在揭示酶的专一性引起的水解产物构象变化与功能特性间的关系,为制备高抗氧化性大豆蛋白乳化剂提供参考。
1 材料与方法
1.1 材料与试剂
大豆分离蛋白(SPI),蛋白质量分数为85.45%;枯草杆菌碱性蛋白酶、菠萝蛋白酶、枯草杆菌中性蛋白酶、胃蛋白酶、胰蛋白酶、十二烷基硫酸钠-聚丙烯酰胺(SDS-PAGE)凝胶制备试剂盒、低分子质量蛋白质Marker等其他试剂均为分析纯。
1.2 仪器与设备
AL204型分析天平,PL303型电子天平,HH-4型数显恒温水浴锅,JJ-1型精密增力电动搅拌器,PHS-3C型雷磁pH计,S22-2型恒温磁力搅拌器,TDL-408型台式离心机,F-4500型荧光分光光度计,Mini-PROTEAN Trtra型垂直电泳槽,MAGNA-IR560型傅里叶红外光谱仪。
1.3 方法
1.3.1 大豆蛋白水解物制备
参照Zheng等[8]的方法略作修改,使用去离子水配制5%(W/V)SPI溶液,于90 ℃加热5 min,酶底物质量比为1∶50,pH值及水解温度设置为酶最适pH值及最适水解温度(具体数值见表1),以保证酶充分发挥作用,水解时间设置为4 h。水解4 h后,水解产物置于90 ℃水浴锅中进行10 min灭酶处理。溶液冷却后调节pH至中性,并于冷冻高速离心机中以10 000×g和4 ℃离心15 min,所得上清液置于冷冻干燥机进行冷冻干燥,既得水解产物。枯草杆菌碱性蛋白酶、菠萝蛋白酶、枯草杆菌中性蛋白酶、胃蛋白酶和胰蛋白酶获得的水解产物分别简称为Alc-SPIH、Bro-SPIH、Neu-SPIH、Pep-SPIH、 Try-SPIH,经杜马斯定氮仪测定,其蛋白纯度分别约为(94.98±0.12)%、(88.73±0.11)%、(86.39±0.04)%、(82.38±0.21)%、(91.84±0.13)%。
1.3.2 蛋白水解度测定
依据NIELSEN等[9]的方法,采用邻苯二甲醛(Ortho-phthalaldehyde,OPA)法进行水解度测定。取冷冻干燥后粉末用磷酸盐缓冲液(0.1 mol/L,pH 7.4)配制为1 mg/mL蛋白溶液,将400 μL上述溶液与3 mLOPA试剂充分混合2 min,并使用光谱仪于340 nm处测定吸光度,每个样品测定3次平行后根据下式计算水解度(DH)。
式中:htot为肽键总数,等于7.8[10];h为水解键数目
1.3.3 溶解度的测定
参考郭荣佳[4]的方法稍作修改进行溶解度的测定。将蛋白粉末溶于去离子水中,使其质量浓度为5 mg/mL,适当稀释后采用微量凯氏定氮法测定上清液蛋白含量。溶解度表示为上清液中蛋白含量占总蛋白含量的比例。
1.3.4 浊度的测定
参照Jean等[11]的方法测定蛋白质浊度,将大豆蛋白水解物稀释成3 mg/mL,用漩涡振荡器混合均匀后,于波长600 nm下测定水解物吸光度,进行3次平行实验后取平均值,得出实验结果。
1.3.5 SDS-PAGE凝胶电泳
参考施蒙[12]的方法稍作调整,将不同酶水解大豆蛋白水解物采用磷酸盐缓冲液(0.1 mol/L,pH 7.4)稀释为2 mg/mL,在25 ℃下孵育1 h后,沸水中煮沸5 min,严格按照分离胶质量分数为12%、浓缩胶质量分数为5%配制电泳胶,上样量为10 μL,保持初始电压80 V约1 h,待样品进入分离胶上沿,调整电压至120 V直至电泳结束。利用固定液染色0.5~1 h后,使用考马斯亮蓝R-250染色3~4 h,随后脱色液进行脱色直至条带清晰。脱色完成后,采用Gel Doc EZ imager型凝胶成像系统分析电泳条带。
1.3.6 疏水性氨基酸浓度测定
采用自动氨基酸分析仪测定疏水性氨基酸浓度。仪器特制水解管内加入20 mg样品及8 mL浓度为6 mol/L的纯盐酸,抽真空条件下封口,于110 ℃条件下水解24 h,待水解完成,用双层滤纸过滤,置于真空干燥器中蒸干并用缓冲液溶解,待仪器测定使用[13]。
疏水性氨基酸浓度=
1.3.7 紫外吸收光谱测定
依据Du等[14]的方法,将样品稀释后使用紫外-可见分光光度计在200~800 nm范围内测定紫外吸收光谱。所有样品均在室温下制备并测定。
1.3.8 荧光光谱测定
参照Sun等[15]的荧光光谱测定方法,样品稀释到质量浓度为2 mg/mL,设置参数为:激发波长为280 nm,发射波长扫描范围为290~450 nm,扫描速度为100 nm/min,激发波长狭缝和发射波长狭缝均为5 nm。每种样品测定3次,所有试验在室温下进行。
1.3.9 傅里叶变换红外光谱测定
利用溴化钾压片法测定傅里叶红外光谱。将冷冻干燥后的大豆蛋白水解物与适量溴化钾充分研磨,实验波数范围为4 000~400 cm-1,分辨率4 cm-1,波数精度0.01 cm-1,扫描次数64次,环境温度25 ℃。利用Peakfit Version 4.0软件对谱带范围1 600~1 700 cm-1拟合,确定蛋白质二级结构的相对含量。以透射率为纵坐标,以波数为横坐标绘图[16]。
1.3.10 乳化活性指数(EAI)和乳化稳定性指数(ESI)测定
参考Molina等[17]的方法稍作修改测定蛋白酶解物样品的乳化活性指数(Emulsifying activity index,EAI)及乳化稳定性指数(Emulsifying stability index,ESI),配制1%蛋白溶液100 mL,吸取75 mL蛋白溶液与25 mL大豆油充分混合,利用高速均质机在10 000 r/min下均质2 min,在0、30 min时分别从烧杯底部吸取20 μL乳状液,加入至5 mL 0.1%的SDS溶液中混合均匀,并在500 nm波长下,以0.1%的SDS溶液为空白样品,测定水解产物吸光值。根据式(1)和式(2)计算EAI和ESI。
(1)
(2)
式中:N为稀释倍数;c为乳化液未形成前蛋白质溶液的质量浓度/g/mL;φ为乳化液中油相体积分数/%;t为间隔时间/min;A0为0 min时吸光度;A30为30 min时吸光度。
1.3.11 ABTS+自由基清除活性
将7 mmol/L的ABTS溶液和2.45 mmol/L的K2S2O8溶液等体积混合并在室温避光条件下静置12~16 h,将混合液以磷酸盐缓冲液(0.2 mol/L,pH 7.4)稀释至734 nm处吸光度为0.70±0.02,吸取4.0 mL上述形成的ABTS+测定液,加入100 μL样品溶液,待充分混匀后测定在734 nm处的吸光值[18]。按式(3)计算ABTS自由基清除率
(3)
式中:A0为ABTS+测定液于734 nm处的吸光度;A1为ABTS+测定液和样品混合后于734 nm处的吸光度;A2为样品于734 nm处的吸光度。
1.3.12 DPPH自由基清除活性
参考Zhou等[19]的方法,配制20 μmol/L的DPPH乙醇溶液并保存在暗处备用。将样品稀释至其吸光值在0.7以下。取1 ml DPPH乙醇溶液与2 mL(1 mg/ml)的样品溶液在暗处混合均匀,充分反应30 min后,测定其在517 nm处的吸光值,平行实验3次。按式(4)计算DPPH自由基清除率:
(4)
式中:AC为DPPH测定液于517 nm处的吸光度;AS为DPPH测定液和样品混合后于517 nm处的吸光度;AB为样品于517 nm处的吸光度。
1.4 数据处理
本实验数据均平行、重复测定3次,采用Excel软件对数据进行统计整理,通过SPSS 24.0进行显著性分析(P<0.05),Peakfitting 4.12软件对红外吸收曲线进行拟合处理,使用origin 2018进行数据的整理与图表的绘制。
2 结果与分析
2.1 水解度、溶解度与浊度分析
蛋白质酶解作用分为轻度酶解和深度酶解,现多用水解度作为产物特性的衡量指标之一。结合蛋白酶不同作用位点可知,如在每种酶的最适pH、最适温度及相同底物特性条件下,酶解程度由大到小依次为:菠萝蛋白酶、枯草杆菌碱性蛋白酶、枯草杆菌中性蛋白酶、胰蛋白酶、胃蛋白酶。表1显示不同酶切方式下水解产物水解度及浊度间差异。Bro-SPIH和Alc-SPIH水解度无显著差异且分别可达到(14.08±0.02)%和(14.07±0.21)%,其主要原因为大豆蛋白在中性及碱性环境中溶解度明显大于酸性环境,且pH位于7.0附近时,大豆蛋白溶解度可达到90%以上,酶与蛋白间相互作用得到极大提升。枯草杆菌中性蛋白酶水解环境同样为中性环境,蛋白溶解度增大,但其作用位点不及菠萝蛋白酶作用位点宽泛,因此水解度为(12.36±0.16)%。胰蛋白酶水解产物Try-SPIH水解度最低,为(3.96±0.02)%,这与江连洲等[20]的研究结果一致,这可能是由于蛋白中胰蛋白酶抑制剂的作用。
溶解度及浊度变化如表1所示。酶解促进蛋白结构展开,由于蛋白质间存在疏水相互作用、二硫键及静电相互作用等作用力,会对酶解后蛋白聚集方式产生影响[12]。浊度可以间接反映蛋白水解物聚集状态。研究表明经过4 h酶解处理产物浊度值均小于SPI浊度值,赵沙沙等[21]指出酶反应到一定阶段后,酶降解蛋白及肽段聚集体速率大于肽聚集速率是浊度下降的主要原因。此外,溶解度情况也是浊度变化的重要原因,如表1所示,SPI溶解度显著低于水解产物溶解度,内部大颗粒物质悬浮,浊度值可达到0.74±0.00。水解产物中Try-SPIH浊度值最大为0.17±0.02,这可能是由于大豆蛋白中胰蛋白酶抑制剂的存在有效抑制蛋白水解,蛋白水解程度及溶解度均较低,光线在溶液中的漫反射程度较大,导致浊度值最大。

表1 不同酶切方式下蛋白水解度及浊度差异
2.2 SDS-PAGE凝胶电泳分析
SPI主要由大豆球蛋白(11S)和β-伴大豆球蛋白(7S)组成,其中11S由酸性亚基A(29~33 ku)和碱性亚基B(22 ku)经过二硫键结合而成,7S则是由α(63 ku),α′(67~72 ku),β(47 ku)3种亚基通过非共价键连接而成[22]。为解析酶解作用效果、水解产物亚基组成,进行SDS-PAGE凝胶电泳实验。图1为SPI及不同酶解条件下蛋白水解物的SDS-PAGE凝胶电泳图。与SPI相比,水解产物均发生不同程度的降解,且组成β-伴大豆球蛋白的α,α′,β这3种亚基均基本被降解,这可能是因为各种蛋白酶优先酶解7S组分[4]。其中Neu-SPIH、Pep-SPIH及Try-SPIH对应于SPI酸性亚基A和碱性亚基B条带处强度均有不同程度降低,Alc-SPIH和Bro-SPIH相应酸性亚基A处条带则彻底消失,生成小分子肽段,这是由于蛋白酶先酶解处于蛋白外部的酸性亚基A,因此位于蛋白质分子内部的碱性亚基B不易进行水解[24]。由图1可得出,枯草杆菌碱性蛋白酶和菠萝蛋白酶水解程度大于其他3种酶类,这也印证了水解度测定结果正确性。
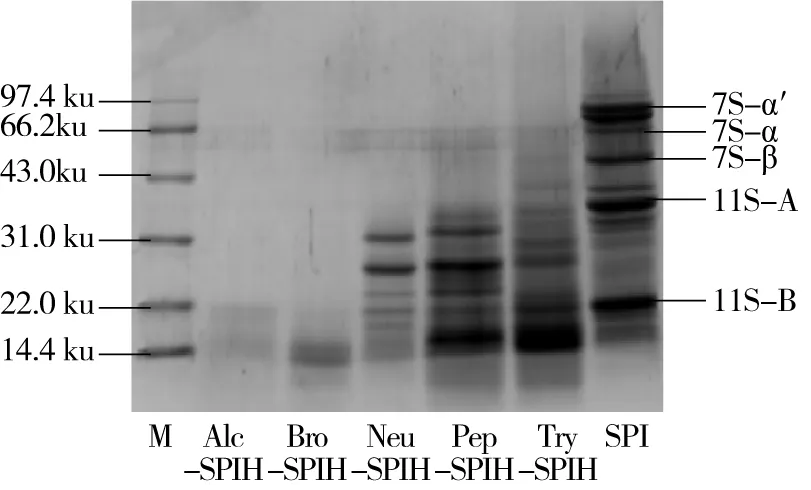
图1 不同酶切方式下蛋白水解物的SDS-PAGE凝胶电泳图
2.3 疏水性氨基酸浓度分析
疏水性氨基酸为侧链具有高疏水性氨基酸的总称。采用自动氨基酸分析仪监测不同酶切方式下疏水性氨基酸浓度变化,用于评估酶作用位点对蛋白氨基酸组成的影响,且与蛋白功能特性密切相关。如图2所示,SPI疏水性氨基酸包裹于球状蛋白内部,较水解样品疏水性氨基酸浓度呈现最小值,这是由于水解产物制备过程中经过离心除去大分子杂质,蛋白溶解度得到显著提升,疏水性氨基酸较SPI得到较大程度保留,从而疏水性氨基酸浓度均高于SPI样品溶液。随着酶切方式的改变,酶特异性作用位点导致酶解作用效果最强的菠萝蛋白酶水解产物疏水性氨基酸质量分数呈现最高值(27.10±0.08)%。枯草杆菌碱性蛋白酶和胃蛋白酶主要水解含有苯环结构或类似苯环结构的芳香族氨基酸形成的肽键,且芳香族氨基酸多为疏水性氨基酸,如苯丙氨酸、酪氨酸、色氨酸,因此这两种酶水解产物中疏水性氨基酸含量较高。胰蛋白酶水解产物疏水性氨基酸所占浓度呈现最低值,可能由两个原因导致,一方面,胰蛋白酶抑制剂限制蛋白展开,水解程度受到抑制,导致疏水性氨基酸比例降低,这可以通过水解度的测定结果来证明;另一方面,胰蛋白酶主要作用于赖氨酸/精氨酸(Lys/Arg)的羧基端,作用位点限制其疏水性氨基酸含量。
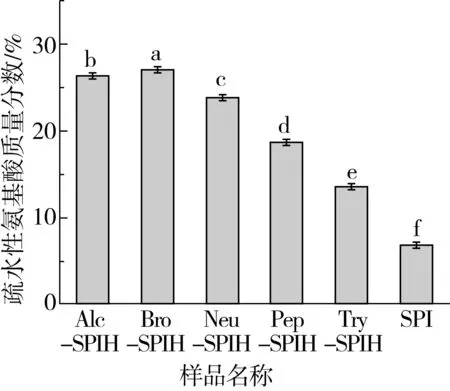
图2 不同酶切方式下蛋白水解物的疏水性氨基酸浓度变化
2.4 紫外吸收光谱分析
图3为SPI及大豆水解产物的紫外吸收光谱图。大豆蛋白的紫外吸收主要来自色氨酸和酪氨酸残基所在侧链基团,其次来自苯丙氨酸、组氨酸、苯丙氨酸残基的侧链[24]。由图3可知,与SPI相比,所有大豆蛋白水解产物峰值均向长波方向红移,表明大豆蛋白空间构象发生变化,蛋白质内部部分分子键断裂,造成特征峰移动[25]。在200~800 nm波长范围内,大豆蛋白具有相似的紫外吸收光谱形状,但水解产物其吸收强度与SPI相比均有明显增强,该结果表明埋藏于分子内部的色氨酸和酪氨酸经蛋白酶作用后暴露于外部[26]。
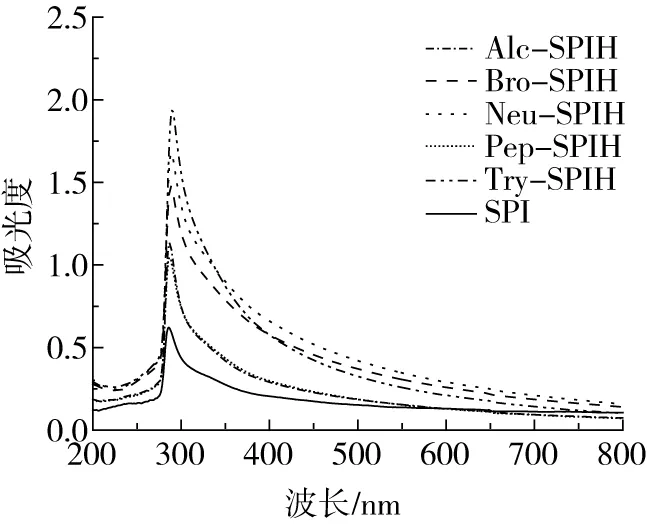
图3 不同酶切方式下蛋白水解物的紫外光谱图
2.5 荧光光谱分析
为探究不同酶切方式对大豆蛋白三级结构的影响,测定SPI及其水解产物荧光光谱变化,如图4所示。大豆蛋白荧光吸收主要来自蛋白内色氨酸残基,因此荧光变化可直接反映色氨酸残基及其微环境变化,进一步确定大豆蛋白质三级结构变化情况[27]。研究表明,最大荧光波长与色氨酸残基存在微环境存在相关性,λmax小于330 nm和大于330 nm分别代表色氨酸残基位于非极性和极性环境中[27, 28]。图4表明,酶解处理后的Alc-SPIH、Bro-SPIH、Neu-SPIH、Pep-SPIH及Try-SPIH的λmax分别为351、346、346、344、339 nm,由此可知不同酶切方式下处理大豆蛋白水解物色氨酸残基均存在于蛋白质极性环境中,表明酶解处理改变蛋白质三级结构及构象,埋藏在内部的色氨酸残基暴露于蛋白质外部环境。与未处理样品比较可知,酶解处理样品荧光光谱峰值向长波长方向发生移动,同时酶解处理均对SPI产生了荧光淬灭,可能是因为酶解促进蛋白结构展开,疏水氨基酸暴露,疏水相互作用增强,导致肽段部分聚集,进而使SPI发生荧光淬灭[21]。
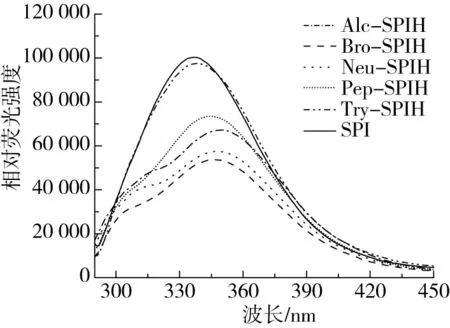
图4 不同酶切方式下蛋白水解物的荧光光谱图
2.6 傅里叶变换红外光谱分析
图5为SPI及不同酶切方式下大豆蛋白水解产物的红外光谱(FT-IR)图。经过不同酶切方式处理,蛋白样品3 305.39 cm-1及1 654.63 cm-1左右的峰宽变窄、峰强增强,可知SPI二级结构发生改变。其中酰胺Ⅰ带(1 600~1 700 cm-1)是大豆蛋白二级结构研究的主要范围,判断红外峰值与二级结构相关关系:1 615~1 637 cm-1和1 682~1 700 cm-1为β-折叠结构;1 646~1 664 cm-1为α-螺旋结构;1 637~1 645 cm-1为无规则卷曲结构;1 664~1 681 cm-1为β-转角结构。确定各样品二级结构相对含量,整理结果见表2。大豆分离蛋白二级结构以β-折叠为主,相对质量分数为(34.22±0.04)%,β-转角质量分数为(23.24±0.04)%,α-螺旋质量分数为(25.63±0.06)%,无规则卷曲质量分数为(16.91±0.03)%。总体而言,酶解处理降低α-螺旋及β-折叠含量,向β-转角及无规则卷曲结构转变。α-螺旋及β-折叠结构中有较多氢键,是维持蛋白刚性结构的重要来源,若二者比例下降,则蛋白柔性结构单元增多,肽段间自由度增大,形成松散有序结构。二级结构变化与酶切位点密切相关,枯草杆菌碱性蛋白酶、菠萝蛋白酶及枯草杆菌中性蛋白酶作用位点主要为疏水性氨基酸残基,且疏水性氨基酸残基主要包埋于蛋白内部α-螺旋及β-折叠结构中,因此其水解产物形成明显柔性结构单元。
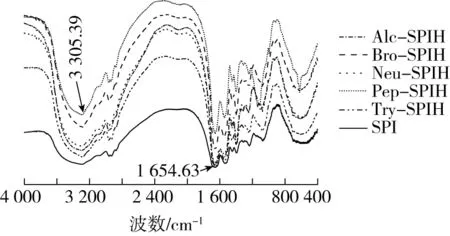
图5 不同酶切方式下蛋白水解物的红外光谱图
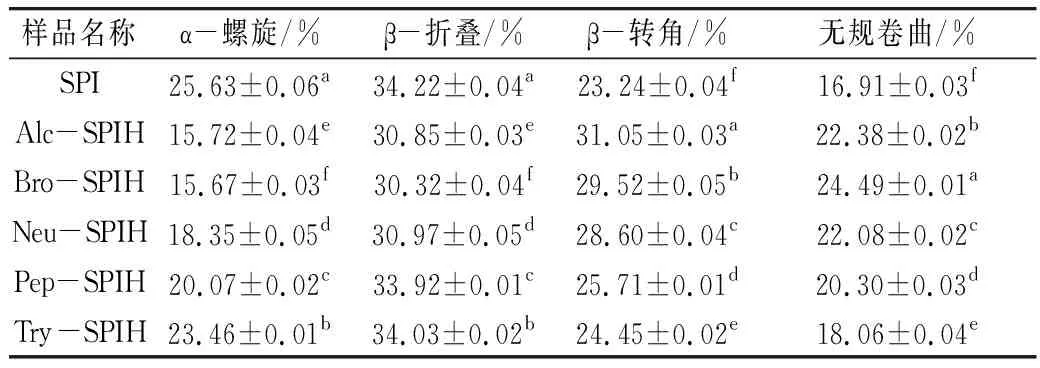
表2 不同酶解蛋白二级结构相对含量
2.7 乳化活性指数(EAI)与乳化稳定性指数(ESI)评价
图6为不同酶解条件对大豆蛋白乳化活性指数(EAI)和乳化稳定性指数(ESI)的影响。EAI值反映了蛋白质在油和水界面吸附的能力,而ESI值则表明蛋白质的稳定性。如图6所示,SPI由于其低溶解度严重限制其乳化性。酶解有效促进SPI构象变化,通过酶切位点的变化,乳化性得到不同程度提升。酶切位点最广泛的菠萝蛋白酶EAI及ESI相较于SPI均显著增加(P<0.05),这主要是因为蛋白内部疏水性氨基酸暴露,电荷数目增加,抑制油滴间相互靠近联结,进而乳化性得到提高。水解程度最低的胰蛋白酶水解产物呈现优异乳化特性[29, 30],主要是因为大豆中胰蛋白酶抑制剂抑制蛋白过度水解,提升蛋白溶解度同时,肽段长度适宜稳定包裹油滴,因此其EAI及ESI均满足乳液要求。

图6 不同酶切方式对蛋白乳化活性指数及乳化稳定性指数的影响
2.8 ABTS+与DPPH自由基清除活性评价
ABTS自由基有机溶剂呈无色状态,易被各种试剂氧化,生成蓝绿色自由基阳离子ABTS+并通常在734 nm处有最大吸收峰,广泛用于抗氧化活性测定。酶解大豆蛋白样品ABTS+自由基清除率如图7所示。大豆分离蛋白ABTS+自由基清除率为(35.30±0.05)%,各酶解产物自由基清除率均高于SPI。结果表明,酶处理可有效提高大豆分离蛋白ABTS+自由基清除率,增强抗氧化活性。这种作用可能归因于酶解对蛋白质结构的影响,蛋白酶促进蛋白质解折叠,形成柔性结构单元,为蛋白酶提供更多作用位点,导致蛋白质分解成肽段。各水解产物ABTS+自由基清除率的差异可能来源于酶作用位点和水解度的影响。部分酶酶解释放疏水性氨基酸残基,改变溶液微环境,不易于水溶性ABTS反应,导致ABTS+自由基清除率较低。DPPH溶液稳定状态呈紫色,遇抗氧化剂转变为黄色,并于517 nm处有最大吸收峰。SPI的DPPH自由基清除率为(61.99±0.07)%,各水解产物DPPH自由基清除率相似且无显著差异。推测原因是蛋白酶切割位点的影响,造成具有DPPH自由基清除活性肽段的释放。
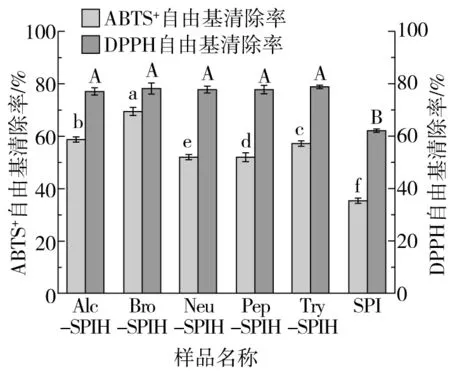
图7 不同酶切方式对蛋白ABTS+与DPPH自由基清除活性的影响
3 结论
本实验比对SPI与不同酶切方式下蛋白水解度、溶解度、浊度、SDS-PAGE凝胶电泳、疏水性氨基酸浓度、紫外吸收光谱、荧光光谱及傅里叶红外光谱间的差异,并针对乳化性及抗氧化性进行评价,探讨酶的专一性引起的水解产物构象变化与功能特性间的关系。结果表明:未处理及不同酶切处理条件下大豆蛋白溶解度、浊度、SDS-PAGE凝胶电泳均随水解度变化相应改变;氨基酸分析发现,酶解后疏水性氨基酸浓度随酶解程度逐渐增大,且均高于SPI内疏水性氨基酸浓度;紫外吸收光谱及荧光光谱进一步佐证酶解作用位点及酶解对微环境改变的影响;傅里叶红外光谱分析则表征蛋白水解物呈现柔性结构,对发挥功能特性提供依据;菠萝蛋白酶水解产物由于其结构展开,疏水性基团暴露,形成柔性结构单元,其乳化性及抗氧化性都得到明显改善。
猜你喜欢
山西化工(2022年6期)2022-10-09
绿色科技(2022年8期)2022-05-25
中国饲料(2022年5期)2022-04-26
药学与临床研究(2021年3期)2021-07-13
农产品加工(2020年17期)2020-10-22
食品安全导刊·中旬刊(2020年2期)2020-06-01
绿色科技(2018年20期)2018-12-19
文苑(2018年22期)2018-11-19
意林·全彩Color(2018年9期)2018-10-12
筑路机械与施工机械化(2017年6期)2017-07-10
