
DSP基因突变致皮肤脆性-羊毛状发综合征1例并文献复习
2022-08-02 02:54汪慧君林志淼
皮肤性病诊疗学杂志 2022年3期
汪慧君, 林志淼
南方医科大学皮肤病医院,广东 广州 510091
遗传性皮肤脆性疾病是一组由皮肤结构异常导致的皮肤黏膜在外力作用下容易出现水疱或缺损的疾病,可伴有毛发指甲等皮肤附属器异常或心脏等其他系统症状[1]。2019年最新的分类标准将其划分为大疱性表皮松解症、表皮剥脱综合征、糜烂性皮肤脆性病、角化过度伴皮肤脆性病及结缔组织疾病合并皮肤脆性病几大类[1]。该组疾病多由角蛋白或细胞间连接蛋白基因突变,导致蛋白数量减少或结构紊乱引起。本文报道1例以皮肤脆性增加、毛发异常伴先天性厚甲的病例,经二代测序结合Sanger验证确定本例患者的致病基因为编码桥粒斑蛋白的DSP基因,因此将患者明确诊断为皮肤脆性-羊毛状发综合征。本研究对探索DSP基因突变与表型的关联性提供了新的依据。
1 对象与方法
1.1 研究对象
患儿男,9个月,出生后不久头部及躯干、四肢部位偶尔出现水疱,皮肤摩擦后易破损。多个指(趾)甲末端逐渐变黄增厚,头发、眉毛无明显生长。皮肤科检查:患者头部、腿部皮肤存在多处散在破溃结痂(图1A、1B),头发、眉毛、睫毛细软稀疏(图1A),口角皲裂、可见黄色痂屑(图1C)。脚趾皮肤轻微脱屑、数个脚趾指甲变黄增厚(图1D)。未见明显掌跖角化。患者身高67 cm,体重7 kg,均低于正常值-2SD。患儿心脏彩超及心电图检查未发现异常改变。父母非近亲结婚,家族内无类似病史。

图1 患者毛发稀疏、短小、细软,头皮多处破损结痂(1A),小腿皮肤破损结痂(1B),口周糜烂、伴有黄色痂屑(1C),脚趾甲末端变黄增厚(1D)
1.2 方法
1.2.1 基因组DNA提取 抽取患者及父母外周血2 mL置于EDTA抗凝管中,用天根血液基因组DNA提取试剂盒(DP349)提取基因组DNA,利用nanodrop微量分光光度计检测DNA浓度及质量。
1.2.2 基因芯片筛查致病基因 将3 μg先证者基因组DNA送往北京迈基诺基因科技股份有限公司,利用GenCap遗传皮肤疾病基因捕获探针V3.0基因检测芯片进行高通量测序,覆盖目标区域长度为2.46 Mb,涵盖726个遗传性皮肤病相关致病基因,除6个基因覆盖全长基因外,其余主要覆盖外显子区域。
1.2.3 Sanger测序验证 针对候选基因突变位点设计引物进行家系共分离验证。用primer-blast在线设计软件(网址:http://www.ncbi.nlm.nih.gov/tools/primer-blast/)针对突变位点所在区域进行特异性引物设计,引物由北京天一辉远生物科技有限公司合成。以家系3名成员的DNA为模板进行目的片段PCR扩增,扩增条件为: 94 ℃预变性5 min后进行以下10个循环:94 ℃变性30 s,62 ℃退火30 s(每个循环退火温度降低0.5 ℃),72 ℃延伸40 s;接着进行25个循环:94 ℃变性30 s,57 ℃退火30 s,72 ℃延伸40 s;最后72 ℃延伸10 min。扩增产物经1.5%琼脂糖凝胶(含GelRed染料)电泳确认大小无误后送往北京天一辉远生物科技有限公司进行Sanger测序。测序结果经序列比对确定有效变异,挑选符合家系共分离的变异位点。
2 结果
患者本人二代测序平均测序深度为513.84,10×覆盖度为99.26%,20×覆盖度为98.9%。经检测发现患者携带19个基因的可疑致病突变,这些基因包括DSP、MBTPS2、KRT16、TRPV3、FANCD2、TYRP1、GGCX、KRT4、HYAL1、SLC27A4、POFUT1、TRIM37、ANKRD11等。经家系验证仅发生在DSP基因的复合杂合突变符合家系共分离,其中c.3805C>T可导致编码的1 269位氨基酸由精氨酸变为终止密码子(p.R1269*),来源于父亲;7568_7571delAGAC突变可导致移码改变(p.T2524Afs*36),来源于母亲(图2A)。根据桥粒斑蛋白的结构组成,两个突变均可导致其编码的桥粒斑蛋白C端结构域出现部分或全部缺失(图2B)。结合患者临床表现及基因检测结果,诊断为皮肤脆性-羊毛状发综合征。
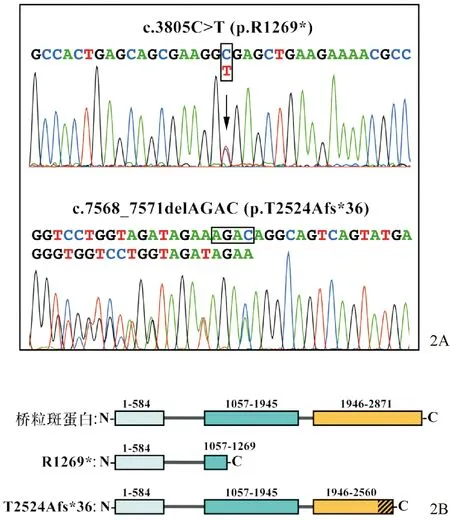
图2 患者DSP基因存在c.3805C>T(p.R1269*)及c.7568_7571delAGAC(p.T2524Afs*36)复合杂合突变(2A)。桥粒斑蛋白全长结构由氨基端结构域(1-584位氨基酸组成)、卷曲螺旋杆状结构域(1 057-1 945位氨基酸组成)及羧基端结构域(1 946-2 871位氨基酸组成)组成,R1269*及T2524Afs*36突变预测可导致蛋白发生部分功能结构域缺失(2B)
3 讨论
桥粒是细胞间连接的关键结构,大量表达于表皮及心肌等承受机械压力的组织[2]。在表皮中,桥粒作为在连接角质形成细胞内中间丝网络与细胞膜的桥梁发挥关键作用[3]。因此,桥粒结构异常不仅影响细胞间连接能力,而且会降低细胞骨架稳定性,导致细胞承受压力能力减弱,引起皮肤脆性增加。自1997年发现首个人类桥粒蛋白——编码桥粒斑菲素蛋白的PKP1基因发生隐性功能缺失突变后可导致外胚层发育不良皮肤脆性综合征[4]以来,国际上陆续发现多种桥粒组成蛋白如桥粒斑蛋白(DSP基因编码)、斑珠蛋白(JUP基因编码)、桥粒芯蛋白1和4(DSG1和DSG4基因编码)、桥粒胶蛋白2和3(DSC2和DSC3基因编码)及角质桥粒蛋白(CDSN基因编码)突变后可导致累及皮肤、毛发或心脏的疾病[5],说明连接蛋白对于维系皮肤及附属器的结构完整性及发挥正常功能具有重要作用。
DSP编码的桥粒斑蛋白是桥粒中含量最为丰富的成分,可变剪切导致其产生两个亚型(DP-Ⅰ和DP-Ⅱ),DP-Ⅰ主要在心脏表达,而DP-Ⅱ主要表达于皮肤[6]。桥粒斑蛋白由中间的杆状结构域及两侧的氨基端和羧基端结构域组成,其中杆状结构域参与二聚合体化,氨基端通过结合斑珠蛋白控制其与桥粒斑块的关联,在组织桥粒钙黏素-斑珠蛋白复合体中发挥作用,羧基端则与胞内的角蛋白中间丝相连,通过连接跨膜钙黏素与胞内角蛋白中间丝网络在表皮和肌肉细胞粘附中发挥主要作用[7]。
DSP基因发生显性错义突变大多引起心律失常性心肌病,如心律失常性心肌发育不良[8-9],少数情况下还可引起严重皮炎-多重过敏-代谢紊乱综合征[10]。1999年Armstrong等[11]报道了首例因为DSP突变引起单倍体效应不足导致的常染色体显性皮肤疾病——条纹状掌跖角化,患者仅出现掌跖角化而无毛发和其它皮肤外表现,这可能是因为承压力较大的掌跖部位需要更多、更高质量的桥粒蛋白以维系其功能,而身体其他部位的皮肤尚可以耐受半量的减少。2000年有研究团队报道了常染色体隐性遗传的DSP突变病例,患者除条纹状掌跖角化外还出现羊毛状发、左心室扩张性心肌病表现,患者所携带的纯合DSP基因截短突变导致编码的桥粒斑蛋白缺乏部分羧基端结构域[12]。有研究曾对1例携带无义及错义复合杂合突变的患者皮损组织进行免疫组化染色,发现突变后的桥粒斑蛋白除了细胞边界定位,还出现了胞浆定位。电镜发现皮损部位棘层松解、细胞间连接减少,角蛋白丝网络的核周凝聚[7],这些发现说明虽然某些情况下机体可以耐受单倍体不足,但如果合并其它突变就会引起更为严重的胞内角蛋白丝结构异常从而引发更为广泛严重的表型。总结文献报道,DSP突变导致的隐性遗传病例主要表型可包括皮肤脆性增加、羊毛状发或脱发、掌跖角化或其他部位角化、厚甲或甲营养不良、心脏异常等,但因累及范围及严重程度差异很大,根据疾病的临床特征组合将其命名为Carvajal综合征、皮肤脆性-羊毛状发综合征及致死性棘层松解型大疱性表皮松解症等疾病[7, 13-17]。本例患者出现皮肤脆性增加、毛发稀疏短小细软及厚甲表现,尽管没有发现明显羊毛状发,但考虑到患者目前不满1岁,毛发尚未发育完全,不排除今后出现羊毛状发的表型,仍将其诊断为皮肤脆性-羊毛状发综合征。
本例患者同时携带一个无义突变和一个移码突变,尽管患者携带的两个突变均造成翻译提前终止,但是由于突变发生在蛋白尾端,结合曾经报道的附近无义突变位点的功能学研究,推测本研究中两个位点导致无义突变介导mRNA降解的概率较低,产生截短蛋白的可能性较大,其中c.3805C>T(p.R1269*)突变会导致翻译提前终止,形成一个缺少大部分卷曲螺旋杆状结构域和全部羧基端结构域的蛋白,而来源于母亲的p.T2524Afs*36移码突变,由于导致读码框发生改变,使得2 524位氨基酸发生改变,并继续翻译出一小段由36个其它氨基酸组成的多肽结构。两个突变均影响了蛋白的羧基端结构域,由于羧基端主要用于与角蛋白中间丝网络相结合,预测可能会影响角蛋白中间丝与桥粒斑块的锚定,造成细胞骨架结构的不稳定。此外,由于卷曲螺旋杆区域与二聚体形成有关,p.R1269*突变还可能会影响到其与T2524Afs*36突变体二聚体结构的形成。上述预测的结构改变解释了患者为何出现皮肤脆性增加的原因。p.R1269*突变位点曾被报道于1例显性遗传的心律失常家系[18],本例患者目前尚未检查到心脏异常,但需要定期检查以防止后期出现心肌改变。
DSP突变导致的疾病表型需要与PKP1基因突变引起的外胚叶发育不良/皮肤脆性综合征相鉴别,PKP1编码的桥粒斑菲素1是一种主要的桥粒装配斑蛋白,可与桥粒斑蛋白1结合,并同样对于角蛋白中间丝的锚定起作用[19]。携带PKP1隐性突变的患者可出现皮肤脆性综合征及累及毛发和指甲等外胚叶发育不良的表型[4, 20],与DSP隐性突变引起的表型高度重合,需要筛查致病基因以明确诊断。
综上所述,本研究报道1例DSP基因突变导致的皮肤脆性增加伴羊毛状发综合征的病例,患者表现出皮肤脆性增加、毛发异常伴先天性厚甲的不典型特征。该病例报道增加了DSP基因突变导致的临床表型谱,有助于提高临床医师的疾病诊断水平,并为探索DSP基因突变与表型的关联性提供新的依据。
猜你喜欢
中国科技财富(2022年8期)2022-12-18
纺织报告(2022年5期)2022-11-22
中国畜牧杂志(2022年1期)2022-11-06
湖北农业科学(2022年11期)2022-07-18
河北农业大学学报(2022年2期)2022-04-26
毛纺科技(2021年3期)2021-04-06
中国麻风皮肤病杂志(2021年2期)2021-01-02
实用肿瘤学杂志(2020年4期)2020-12-08
中国航海(2019年3期)2019-10-30
北京航空航天大学学报(2017年12期)2017-04-23
