
Alport综合征合并Gitelman综合征1例报告
2022-08-03 11:27吕磊阳曹青洋
长治医学院学报 2022年3期
王 鑫 吕磊阳 王 倩 曹青洋 李 俊
Alport综合征(Alport syndrome,AS)和Gitelman综合征(Gitelman syndrome,GS) 是遗传性肾脏疾病,两者确诊都需要依靠基因检测。本文将报道1例通过基因检测发现变异基因COL4A5基因及SLC12A3基因,同时患有Alport综合征和Gitelman综合征的病例。
1 临床资料
患儿,男,8岁,主因“发热4 d”于2021年3月收入我院儿科。既往史:患儿2014年曾因“血尿”住院,考虑为“急性肾炎”,经治疗好转出院,出院后曾口服“中药”治疗,未规律复查。家族史(见图1):外祖母有2个弟弟,年幼时因病去世,具体病因不详。外祖母(Ⅰ2)、表外祖母(Ⅰ3)、舅舅(Ⅱ1)、表姨(Ⅱ6、Ⅱ8)及表姨家女儿(Ⅲ3)、儿子(Ⅲ5)患有“肾病”,舅舅现行“血液透析”治疗,表姨家儿子、女儿二人听力下降,均已行“肾移植”治疗。母亲(Ⅱ4)自诉孕期检查尿常规有“潜血”,未诊治。该家族中所有患者都未行肾脏活检或相关基因检测。
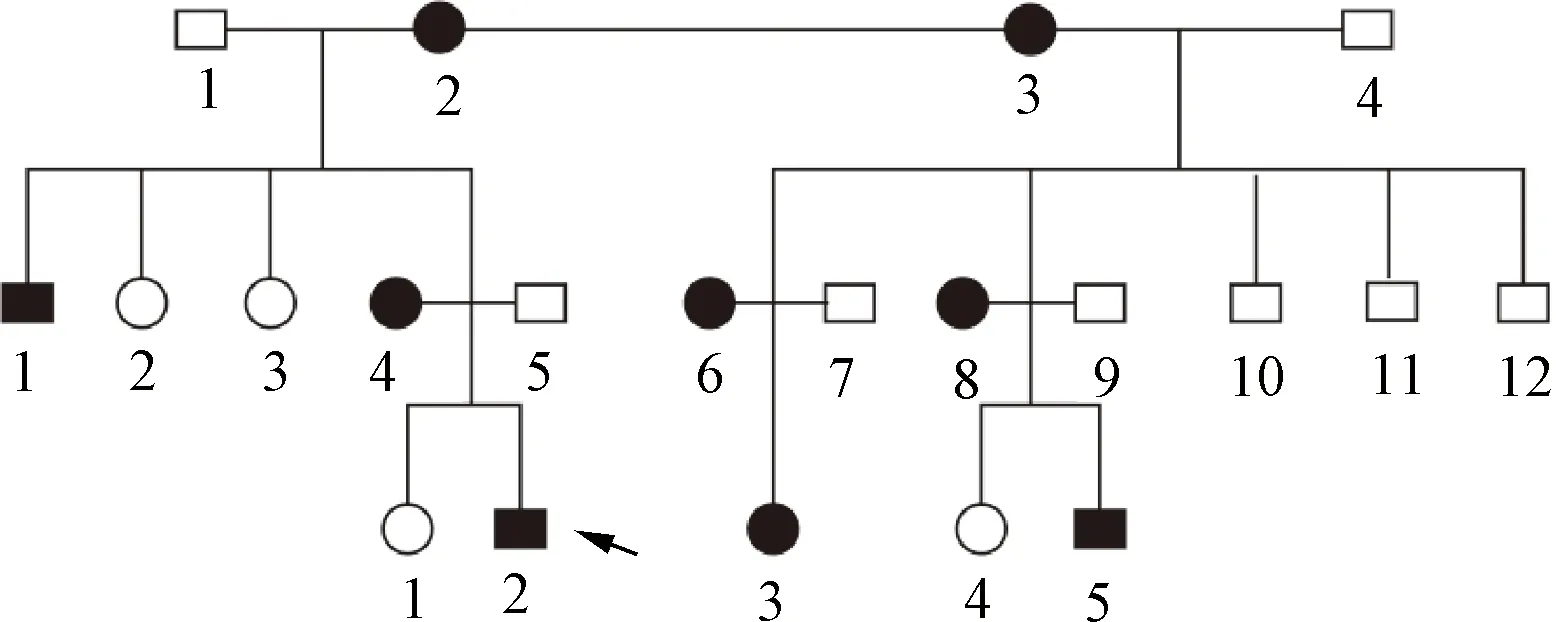
代表男性,○代表女性,●代表患有“肾病”者,箭头示患儿图1 Alport综合征家系图
体格检查:体温37.5 ℃,心率90次·min-1,呼吸18次·min-1,血压90/56 mmHg,身高120 cm,体质量20 kg,神志清楚,精神一般,左侧眼裂较右侧略小,双眼略突出,无眼睑浮肿,咽部略充血,双肺呼吸音粗,未闻及干湿性啰音,心音有力,律齐,心脏各瓣膜听诊区未闻及杂音,腹软,肝肋下未触及,肾区无叩击痛,神经系统未见异常。
辅助检查,血细胞分析:白细胞3.33×109L-1,中性粒细胞百分比73.3%,淋巴细胞百分比17.8%;C-反应蛋白55.24 mg·L-1,降钙素原0.67 ng·mL-1,红细胞沉降率:75 mm·h-1,电解质:钾3.12 mmol·L-1,钙 1.95 mmol·L-1;肺炎支原体抗体检测:1∶320,尿有形成份分析:尿蛋白2+,潜血3+,白蛋白肌酐比2+,镜检:红细胞满视野/HP;超高倍尿液细胞形态分析:红细胞满视野/HPF,混合性非均一,小红细胞/皱缩红细胞,50%<畸形率<70%。24 h尿蛋白:0.184 g,24 h尿钙:0.24 mmol。肾功能、肝功能、总胆固醇、甘油三酯、乙肝五项定性、免疫球蛋白IgA、IgG、IgM、单项补体、抗链球菌溶血素O测定、中性粒细胞胞浆抗体、抗双链DNA抗体、抗核抗体、类风湿因子、甲功五项、血培养均未见异常。胸部正位片、心电图、左肾静脉彩超、泌尿系彩超:未见明显异常。普通测听纯音:双耳感音神经听力下降,声导抗未见明显异常。左眼视力0.8,右眼视力0.5,双眼前节(-),眼底无异常。基因分析:泌尿系统疾病Panel V4(肾病整体解决方案),(1)发现COL4A5基因有1个半合子突变(见图2、图3),c.3152 G>A患儿母亲该位点杂合变异,在3152号核苷酸由鸟嘌呤 G变为腺嘌呤A(c.3152 G>A)的半合子突变,导致第1051号氨基酸由甘氨酸变为谷氨酸(p.G1051E);(2)发现SLC12A3基因有1个杂合突变(见图4),外显子6(c.841 T> C)发生杂合突变,在281号密码子(p.W281R) 上引起精氨酸取代了色氨酸。

图2 患儿COL4A5基因变异图

图3 患儿及母亲COL4A5基因分析结果图

图4 患儿SLC12A3基因变异图
2 诊疗
入院时给予头孢呋辛抗感染、补液纠正电解质紊乱治疗,于入院第5天,因患儿反复发热,支原体抗体阳性,考虑肺炎支原体肺炎导致发热,同时考虑存在Alport综合征,加用阿奇霉素抗感染、口服雷米普利片治疗,入院第8天患儿体温恢复正常,复查尿有形成份分析:尿蛋白 2+,潜血 3+,镜检:红细胞满视野/HP;超高倍尿液细胞形态分析: 红细胞满视野/HPF, 较入院时变化不大。电解质:钠136.1 mmol·L-1、钙2.0 mmol·L-1。入院第10天,由于患儿一般情况好,临床表现仅小便为褐色,家属要求出院,考虑患儿存在Alport综合征合并Gitelman综合征,为明确患儿SLC12A3基因c.841 T>C杂合变异的来源,建议父母检测SLC12A3基因c.841 T>C变异,被家长拒绝。院外予以雷米普利片1 mg,每日1次口服,嘱咐多进食含氯化钠、含钾、镁丰富的食物,并定期复查电解质、血镁、尿钙、尿常规。由于患儿及家属依从性差,仅在出院后2个月进行1次复查,尿有形成份分析:尿蛋白1+,潜血3+。近期随访中家属表示患儿目前一般情况可,无异常表现,未规律服用药物。
3 讨论
AS是一种遗传性进行性肾病,发病率约为1/50 000[1],以血尿、感音神经性耳聋、特殊的眼部异常(圆锥形晶状体和黄斑斑点)、进行性肾功能减退、终末期肾脏病为临床典型表现[2]。该病是由肾脏基底膜(GBM)IV 型胶原蛋白网络缺陷引起的。AS约85%的病例是由IV型胶原α5链的COL4A5基因突变引起的的X连锁显性遗传型[3],约有15%的病例是由α4链的COL4A4基因和α3链的COL4A3基因突变引起的常染色体隐性方式遗传型[4],在非常特殊的情况下可表现为显性遗传[5]。病初光学显微镜下的肾脏活检标本可能是正常的,随着疾病的进展,可能会出现非特异性的表现,包括局灶性和节段性肾小球硬化,肾小管萎缩,间质纤维化以及间质浸润。免疫荧光检查多为阴性结果[6]。电子显微镜(EM)可以发现肾小球基底膜的不规则增厚或变薄,致密层劈裂、分层、篮网状改变[7]。
除外X连锁AS的女性中血尿的可能性为95%[8],其余所有AS患者都有持续性血尿特征,因此尿液分析是一种非常有效的AS筛查方法[9]。在有些国家,首选的检测方法为肾脏活检,但肾组织电镜下的典型改变仅见于约60%的AS患者[10],且对AS的预后没有价值,因此在欧洲,医生更喜欢通过基因检测来确诊[11]。基因检测作为AS诊断的确诊手段比肾脏活检更具有敏感性、特异性且有重要的预后价值,尽早进行基因检测显得尤为重要。在近期的AS分子诊断会议中将筛查COL4A5、COL4A3和COL4A4基因致病变异的适应症扩展到了典型AS表现(血尿、肾功能衰竭;血尿或肾功能衰竭家族史)之外,包括持续性蛋白尿、类固醇抵抗性肾病综合征、局灶节段性肾小球硬化(FSGS)、家族性IgA 肾小球肾炎和无明显原因的终末期肾功能衰竭[12]。
GS是SLC12A3基因发生功能缺失或突变导致肾远曲小管的噻嗪类利尿剂敏感的钠氯共同转运体(NCCT)蛋白的结构和(或)功能异常,从而引起肾脏远曲小管对钠氯重吸收障碍[13]。GS很少见,估计发病率为1/40 000[14]。大多数患者临床表现为乏力、多尿、口渴、生长发育落后等非特异性表现,除外偶然诊断,它通常在儿童晚期或成年早期被诊断出来,而在这些报告中多数是通过临床诊断并非是基因检测基础上诊断[15]。
根据BLANCHARD A等[16]发表的“改善Gitelman综合征预后的共识和指导意见”和2017年的“Gitelman综合征诊治专家共识”[17],将SLC12A3基因中发现2个致病突变作为GS的确诊标准,但在许多具有GS的临床和生化特征的患者仅携带1个致病突变[15]。
目前尚无彻底治愈AS的方法,临床上多采用减少尿蛋白、抗纤维化等对症治疗以延缓肾功能进一步恶化。KASHTAN等[18]阐述了血管紧张素转换酶抑制剂(ACEI)作为治疗一线的用药,血管紧张素受体阻滞剂(ARB)为二线用药。现在越来越多的证据表明,早期应用ACEI可显著改善AS的肾脏结局,在2020年KASHTAN等[11]在新的治疗建议中阐述了早期特别是在肾功能下降之前应用ACEI对AS治疗的重要性。随着基因技术的不断发展,基因治疗逐渐成为AS治疗领域的热点,而且已经取得有意义的研究进展,如NOZU等[19]正在开发使用反义寡核苷酸(ASO)的外显子跳跃疗法,以治疗严重的男性X 连锁显性病例,他们正在使用动物进行安全性评估测试,并将进行临床试验,这使将来基因治疗用于临床成为可能。
GS治疗主要使用高钾、高镁、高钠替代品,效果不好或不能耐受副作用时,可考虑使用潴钾类利尿剂、肾素-血管紧张素拮抗剂或非甾体消炎药(NSAID),甚至上述药物联合应用[16-17]。
在本病例中,患儿入院时以“发热”为主要表现,查体无明显阳性体征,化验外周血白细胞计数、中性粒细胞比例增高,故入院时考虑为上呼吸道感染。在尿常规检查中发现“尿蛋白2+,潜血3+”,超高倍尿液细胞形态分析:红细胞满视野/HPF,再次向家属仔细了解既往史及家族史,得知患儿家族中有“肾脏”疾病遗传史,故考虑诊断:Alport综合征可能。在检查中因患儿家属拒绝进行肾穿刺活检,故缺少了AS电镜下肾小球基底膜的超微改变的依据,但在基因分析中发现COL4A5基因有1个半合子突变,c.3152 G>A患儿母亲该位点杂合变异。结合患儿病例特点,符合AS诊断标准。在该基因分析报告中,另外发现SLC12A3基因1个致病突变,该患儿身高、体重均低于同年龄、同性别2SD,存在发育迟缓,存在低钾血症、低尿钙,肾脏超声正常,可以诊断该患儿患有GS。
AS和GS皆为肾脏疾病,尚不清楚两者同时存在是否会导致肾脏损伤加重。两者在治疗方面存在差异,但也存在共同点,即使用肾素-血管紧张素拮抗剂治疗,且ACEI类药物优先于ARB类药物[11,16-17]。目前在治疗上尚未对两者同时存在有明确的建议。
本病例强调当有孤立性肾小球血尿患者且有血尿家族史或慢性肾脏病史、双侧耳感音神经性听力损失或眼部异常的患儿应行AS相关基因检测明确诊断。GS多数为青年或成年发病,且临床表现是非特异性的,可在儿童期出现临床症状,多与电解质紊乱及肾素-血管紧张素-醛固酮系统(RAAS)激活有关,当我们在临床工作中遇到这种非特性的表现,应及时行相关实验室检查及基因检测。
猜你喜欢
现代临床医学(2022年1期)2022-02-12
祝您健康·文摘版(2021年12期)2021-12-08
求学·理科版(2021年3期)2021-07-28
家庭医学(2020年10期)2020-11-20
特别健康·下半月(2020年9期)2020-09-21
祝您健康(2000年11期)2000-12-31
祝您健康(1987年6期)1987-12-31
