
环氧树脂定型羧基化聚乙二醇相变材料的制备及其性能
2022-08-04 08:20孙义明冉宝清王福瑶卞武勋
合成树脂及塑料 2022年4期
孙义明,冉宝清,梅 捷,王福瑶,卞武勋*
(1. 湖北工业大学 材料与化学工程学院,湖北 武汉 430068;2. 湖北工业大学 绿色轻工材料湖北省重点实验室,湖北 武汉 430068)
相变储能材料具有良好的储放热功能,通过材料相态变化来完成能量的储存和释放,具有能量密度高、化学稳定、成本低等优点,广泛应用于建筑保温[1]、食品保鲜[2]、太阳能[3]、废热收集[4]等领域。现有的储热技术可分为显热储能、潜热储能和化学反应储能。显热储能存在能量密度低、变温放热等缺点,因此不是一种良好的储热技术;化学反应储能是利用可逆化学反应来储存能量的一种技术,但是该化学反应的发生需要特定的条件,技术复杂,实用价值不大;潜热储能具有能量密度高,放热过程为等温过程等优点得到了广泛应用,潜热储能是储存能量有效的方法之一[4]。Sari等[5]制备了聚乙二醇(PEG)接枝苯乙烯马来酸酐共聚物。研究表明,随着接枝的PEG含量的变化,相变焓为107.0~155.0 J/g,并且材料在相变温度内发生固固相变。基于同样的原理,Liu Luntao等[6]使用乙烯马来酸酐共聚物与十四醇、十六醇、十八醇等脂肪醇反应,得到的相变材料的焓值为109.0~135.8 J/g。Lu Xiang等[7]采用三聚六亚甲基二异氰酸酯(HDIT)与PEG在无溶剂的情况下制备了定型相变材料,通过向PEG中加入HDIT来控制交联密度,w(HDIT)为10%时,相变焓为89.7 J/g,但该体系只能依靠两者的投料比来控制交联密度,HDIT含量过多或过少时,该反应有可能无法生成凝胶。Zhou Xiaoming[8]使用二苯基甲烷二异氰酸酯(MDI)将PEG作为侧链接枝在聚乙烯醇(PVA)分子链上制备了定型相变材料,其熔融焓为72.8 J/g,此研究使用的PEG,MDI和PVA都是多官能度化合物(官能度大于等于2),因此是一个可以生成凝胶的体系;但由于MDI与PVA,PEG的反应存在许多不可控因素,整个反应体系存在很多副反应(如PVA之间的交联,PEG自身发生扩链),这些副反应可能会导致所制相变材料的结构与预期结构存在差异。本工作采用酯化接枝羧基的方法对PEG进行端基修饰,利用官能团活性的差异将形成交联网络的PEG与用作填充的PEG进行区分,前者对环氧树脂有更高的活性,这样在后续实验中就有更大的余地来调节交联密度。同时,由于端基修饰后的PEG可以参与环氧树脂的固化反应,此时,由环氧树脂所形成的定型骨架同样具有一定的相变焓,避免了骨架不参与储能过程而导致相变焓降低。
1 实验部分
1.1 主要原料与仪器
乙二醇二缩水甘油醚(EGDE),环氧值大于0.7 mol/100 g;1,2,4-偏苯三酸酐(TMA);PEG4000(4 000代表PEG的相对分子质量,下同),PEG20000:均为化学纯,麦克林生化科技有限公司。高纯氮气,纯度99.9%,武汉市博强气体有限公司。
TensorⅡ型傅里叶变换红外光谱仪,Ascend400型核磁共振波谱仪:德国Bruker公司;PL-GPC50型凝胶渗透色谱仪,美国Agilent公司;Empyrean型X-射线衍射仪,荷兰帕纳科仪器公司;DSC 8000型差示扫描量热仪,美国PE公司。
1.2 试样制备
1.2.1PEG羧基化
将PEG4000于80 ℃干燥12 h后转移至三口烧瓶中,升温至120 ℃,通过鼓泡器控制氮气流量为5 mL/s;5 min后,向其中加入计量的TMA,继续反应一定时间,期间保持氮气流量为5 mL/s,得到TMA接枝PEG4000的产物(PEG4000TMA),反应机理见图1。
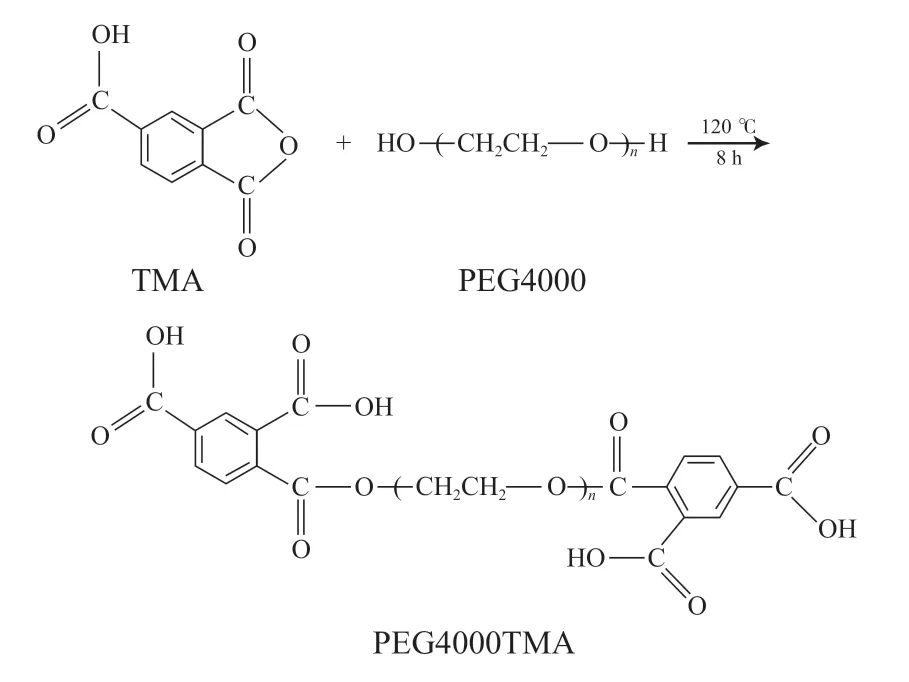
图1 PEG4000羧基化反应示意Fig.1 Schematic diagram of PEG4000 carboxylation reaction
1.2.2定型相变材料的制备
将11.06 g PEG4000TMA转移至烧杯中,加入3.84 g的EGDE,搅拌均匀,倒入聚四氟乙烯模具中,温度105 ℃,4 h完成固化,产物为PEG定型相变材料(记作SS-PCM1),交联示意见图2。
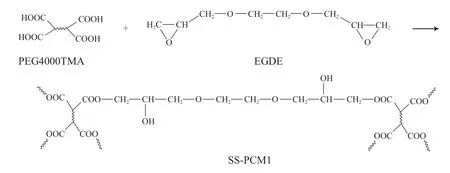
图2 羧基化PEG交联过程示意Fig.2 Schematic diagram of PEG crosslinking process
将11.06 g PEG4000TMA转移至烧杯中,加入11.06 g的PEG20000搅拌均匀,再加入3.84 g EGDE,搅拌均匀,倒入聚四氟乙烯模具中,温度105 ℃,4 h完成固化,产物记作SS-PCM2。
1.3 测试与表征
傅里叶变换红外光谱(FTIR)分析:取少量试样用丙酮溶解,配制成稀溶液,将配制好的溶液滴到KBr压片上,置于干燥箱,待丙酮溶剂完全挥发后进行测试,波数为400~4 000 cm-1。
核磁共振氢谱(1H-NMR)分析:将20 mg试样溶于2.5 mL氘代氯仿中,使其形成均匀溶液后进行测试,内标物为四甲基硅烷,采样时间为5 s,扫描次数大于5 000次。
凝胶渗透色谱(GPC)分析:取PEG4000TMA和PEG4000,以四氢呋喃为流动相,聚苯乙烯为标样,测定相对分子质量及其分布,流量为1.0 mL/min。
X射线衍射(XRD)分析:Cu靶,Kα射线,高压发生器电压为45 kV,电流为40 mA,测试温度为室温,衍射角(2θ)为0°~50°,扫描速率为5(°)/min。
差示扫描量热法(DSC)分析:将3.0~5.0 mg试样密封于坩埚,氮气气氛,测试温度为0~100 ℃,升降温速率均为10 ℃/min。
2 结果与讨论
2.1 FTIR分析
从图3可以看出:PEG4000谱线中,1 090 cm-1处为C—O的伸缩振动峰,2 886 cm-1处为C—H的伸缩振动峰,2 543 cm-1处为游离态O—H的伸缩振动峰,960 cm-1处为PEG的结晶峰。TMA谱线中,1 013 cm-1处为C—O—C的对称伸缩振动峰,1 223 cm-1处为C—C—O的反对称伸缩振动峰,1 649 cm-1处为羧基中C=O的伸缩振动峰,1 796 cm-1处为酸酐的C=O伸缩振动峰,3 091 cm-1处为苯环上C—H的伸缩振动峰,3 247 cm-1处为O—H的伸缩振动峰。PEG4000TMA谱线中,1 796 cm-1处酸酐的C=O伸缩振动峰消失,仅存在1 727 cm-1处羧基中C=O的伸缩振动峰,证明产物中酸酐基团已经消失。

图3 TMA,PEG4000,PEG4000TMA的FTIRFig.3 FTIR of TMA,PEG4000,PEG4000TMA
2.2 GPC分析
从图4可以看出:PEG4000,PEG4000TMA的重均分子量(Mw)分别为4 845,5 821,数均分子量(Mn)分别为4 197,4 395,羧基化导致改性PEG的相对分子质量有一定的增长,相对分子质量分布(Mw/Mn)分别为1.155,1.325。Mw/Mn仅有少许加宽,Mn和Mw都些许增大;但是相较于理论Mn,实际测试得到的Mn偏小。综合分析认为原因是:(1)此反应没有添加溶剂,所以会有部分PEG链端的羟基被包埋,酸酐无法与其接触发生反应。(2)体系中存在游离的TMA或其水解产物,Mn在统计的过程中,相对分子质量小的组分会对其有较大的“贡献”,导致实测Mn偏小。同时,根据PEG4000与PEG4000TMA的相对分子质量变化情况可以得出:在此体系中,基于羧基与羟基直接发生酯化或由于酯交换反应等副反应生成的扩链产物几乎没有。

图4 PEG4000与PEG4000TMA的GPCFig.4 GPC curves of PEG4000 and PEG4000TMA
2.3 1H-NMR分析
从图5可以看出:PEG4000谱线中,化学位移在3.6处为PEG上亚甲基的质子峰,2.4~2.6可能为PEG分子链端羟基上的质子峰,这是由于羟基上的氢较为活泼,会与氘代氯仿中的氘发生置换,使峰的位置发生变化并且使峰形变宽,甚至消失。PEG4000TMA谱线中,化学位移在4.5处为PEG链端亚甲基的质子峰,这个亚甲基由于化学环境发生了变化,因此向低场方向发生了移动;化学位移在7.8处的两个裂分峰为苯环上相邻的两个质子,两个质子之间相互耦合,因此,由单峰变成了裂分峰;化学位移在8.4处为苯环上夹在两个羧基之间的质子。综合得出PEG的端羟基参加了反应。

图5 PEG4000与PEG4000TMA的结构式及1H-NMRFig.5 Structure and 1H-NMR of PEG4000 and PEG4000TMA
2.4 XRD分析
从图6看出:PEG4000谱线中,2θ为19.31°处的衍射峰为(120)晶面,23.45°处的衍射峰为单斜晶胞的(032)晶面[9]。SS-PCM1和SS-PCM2的谱线中,2θ为19.31°,23.45°处也出现了衍射峰,两条曲线的衍射峰位置几乎相同,也没有新的峰出现。这说明PEG经羧基化修饰及引入环氧基团交联后,没有对PEG的晶型造成明显影响。一般认为,PEG的结晶过程包括成核和生长两个步骤,在PEG分子结晶的早期,其聚合物链的热波动引起了晶体的初生核化;随后逐渐生长为晶体。从图6还看出:2θ为23.2°处,SS-PCM1及SS-PCM2的衍射峰明显低于PEG4000,这是由于对部分PEG4000端基进行了修饰,影响了被羧基修饰的PEG4000分子链运动、导致结晶能力下降的缘故。

图6 PEG4000,SS-PCM1,SS-PCM2的XRD图谱Fig.6 XRD specturm of PEG4000,SS-PCM1 and SS-PCM2
2.5 DSC分析
相变潜热是衡量相变储能材料储能能力及是否能够实际应用的重要指标,从图7可以看出:PEG4000的熔融温度为59.73 ℃,SS-PCM1的熔融温度为47.72 ℃,SS-PCM2的熔融温度为56.65℃。PEG4000的结晶温度约为30.00 ℃,SS-PCM1的结晶温度为19.77 ℃,SS-PCM2的结晶温度为36.49 ℃。PEG4000的熔融焓为179.67 J/g,SSPCM1的熔融焓最低,仅有78.33 J/g,SS-PCM2的熔融焓有一定的提升,从SS-PCM1的78.33 J/g上升至126.64 J/g,上升了约61.7%。其原因为:(1)SSPCM1中的PEG4000TMA完成交联之后,链段运动能力受到了限制,需要有更强的推动力才能完成结晶过程,因此SS-PCM1结晶温度较低。这是因为在交联点附近的PEG链段受到了分子链的限制,链段不能正常地排列到相应的晶格中,使材料的结晶度下降导致的,这与许多研究者的结论一致[10-11]。(2)SS-PCM2的结晶温度较SSPCM1有所提高,可能是因为SS-PCM2中PEG含量高于SS-PCM1,受限分子相对较少,更容易结晶。SS-PCM2的结晶温度甚至高于PEG4000,这是因为SS-PCM2中含有大量的PEG20000分子,较PEG4000分子的链段更长,结晶过程中PEG20000的链段可能起到了近似异相成核的作用。(3)SSPCM1与SS-PCM2两种定型相变材料中,只有PEG链段有结晶能力,本工作为了实现定型,向其中加入了环氧树脂和酸酐,这些组分不参与相变储能过程,相当于“稀释”了材料原有的相变焓,导致两种定型相变材料相变焓下降。
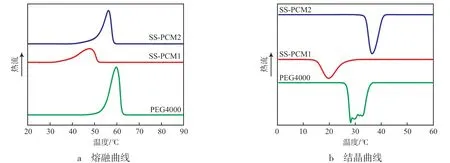
图7 PEG4000,SS-PCM1,SS-PCM2的DSC曲线Fig.7 DSC curve of PEG4000,SS-PCM1 and SS-PCM2
3 结论
a)使用TMA成功完成了PEG的羧基化处理。
b)羧基化导致改性PEG的相对分子质量有一定程度的增长。
c)制备的定型相变材料SS-PCM1的熔融焓较低,为78.33 J/g,通过填充PEG20000可以进一步提升其焓值。
猜你喜欢
中国科技财富(2022年8期)2022-12-18
橡塑技术与装备(2022年8期)2022-12-17
高分子材料科学与工程(2022年8期)2022-11-02
中国农业科学(2022年17期)2022-09-19
物理学报(2022年10期)2022-06-04
科技视界(2022年9期)2022-04-09
天津诗人(2021年1期)2021-11-12
物理实验(2020年12期)2021-01-06
建材发展导向(2020年16期)2020-09-25
疯狂英语·新阅版(2019年6期)2019-09-10
