
83例肝豆状核变性患者的临床特征分析
2022-09-07 04:40赵素贤张庆山孔令波任伟光
临床肝胆病杂志 2022年8期
纪 雷, 张 莹, 孔 丽, 赵素贤, 崔 坡, 张庆山, 孔令波, 任伟光
河北医科大学第三医院 中西医结合肝病科, 石家庄 050051
肝豆状核变性又称威尔逊病(Wilson’s disease,WD),是一种常染色体隐性遗传的铜代谢相关疾病。我国WD患病率为1.96/10万,明显高于欧美国家[1]。WD发病基因ATP7B编码转运P型ATP酶,参与铜的跨膜转运,当其发生基因变异时,铜的转运与代谢发生障碍,过量蓄积于肝、脑、肾等部位,造成多系统损害[2]。该病起病隐匿,临床表现多样,因此早期诊断困难,易造成漏诊误诊。现将本院收治的83例WD患者的临床特征总结如下。
1 资料与方法
1.1 研究对象 回顾性收集2013年4月—2021年8月在本院确诊的WD患者的病历资料。纳入标准:参考《中国肝豆状核变性诊治指南2021》[3],结合肝损伤及椎体外系受损表现,角膜K-F环阳性,血清铜蓝蛋白下降,24 h尿铜升高及影像学表现确诊WD。排除标准:合并自身免疫性疾病或其他原因引起的胆汁淤积性肝病者。

2 结果
2.1 一般资料 共收集WD患者83例,其中男40例,女43例,男女比1:1.075;发病年龄3~63岁,平均(21.16±14.87)岁,其中≤18岁者39例(46.99%)。从发病到确诊的时间为1~37年,中位7年。有明确WD家族史者5例,疑似家族史者2例。83例患者中以肝损伤为首发表现者(肝型)53例(63.86%);以神经系统损伤为首发表现者(脑型)6例(7.23%);同时具有肝脏及神经系统症状者(混合型)18例(21.69%);以骨骼等其他系统损伤为首发症状者(其他型)6例(7.23%)。各临床分型及年龄分布特点见表1。
2.2 临床表现 (1)肝脏受损情况:主要表现为乏力、纳差19例,腹胀、腹痛17例,皮肤巩膜黄染12例,双下肢水肿9例及单纯转氨酶升高5例。临床诊断为慢性肝炎27例(轻度9例、中度15例、重度3例),肝硬化31例(肝硬化合并食管胃底静脉曲张出血8例,肝硬化合并肝性脑病3例),肝衰竭5例(慢加急性肝衰竭4例,暴发性肝衰竭1例),慢性乙型肝炎、慢性丙型肝炎和酒精性肝病合并肝豆状核变性各1例,无症状型5例(2例因姐姐或弟弟确诊WD后发现仅铜蓝蛋白水平下降)。(2)神经系统受损症状:主要表现震颤、口下颌肌张力障碍、运动迟缓及精神异常,其中四肢震颤16例,言语不清、说话慢11例,动作迟缓、走路姿势异常9例,精神异常5例(反应迟钝、注意力不集中、智力下降、学习成绩下降及记忆力下降各1例)。(3)骨关节损伤6例,其中双膝、双踝疼痛4例,双下肢畸形1例,胸骨凹陷1例。
2.3 角膜K-F环及实验室检查 (1)角膜K-F环阳性62例(74.69%),不同临床分型的K-F环结果见表2。(2)铜生化:血清铜蓝蛋白水平下降79例(95.18%),不同临床类型之间血清铜蓝蛋白水平差异无统计学意义(F=1.013,P>0.05);24 h尿铜水平升高73例(87.95%)。不同临床类型之间铜生化特征见表2。(3)血生化:ALT或AST升高52例(62.65%),低蛋白血症30例(36.14%);肌酐水平升高2例(2.41%)。(4)血常规:贫血40例(48.19%),其中轻度33例、中度6例、重度1例。
2.4 头颅影像学检查 17例患者行头颅CT或MRI检查,显示为多发异常信号,累及的部位依次为基底节区8例,脑干、丘脑各4例,豆状核、尾状核各2例,脑白质变性、脑室系统扩张及脑沟、池、裂增宽各1例。
2.5 肝组织病理学检查 25例患者行肝组织病理检查,结果显示,慢性肝炎期12例,主要表现为汇管区炎性细胞浸润、胆管上皮变性、小叶内灶性炎症,伴较多铜颗粒沉积;肝硬化期10例,表现为汇管区扩大纤维化,纤维间隔内铜颗粒沉积及小胆管破坏、增生、间隔周围肝细胞胆盐淤积性改变及铜颗粒沉积;3例肝细胞呈大小泡混合性脂变,伴少量铜颗粒沉积。
2.6 ATP7B基因检测 30例患者行基因检测,其中25例(83.3%)患者检测结果异常(复合杂合突变18例,杂合突变4例及纯合突变3例);共检测到30种不同的ATP7B等位基因突变,包括20种错义突变、6种剪接错误及4种基因多态。最常见的等位基因突变为外显子8的c.2333G>T/p.R778L,等位基因频率为60%;其次为外显子13的c.2975C>T/p.P992L、外显子11的c.2621C>T./p.A874V及外显子16的c.4003G>C/p.V1140A,等位基因频率依次为24%、12%及4%。
3 讨论
WD可累及多个器官,临床表现多样,易漏诊或误诊,尽管基因诊断技术有助于症状前期或产前诊断,但全面认识WD的临床表现、体征及辅助检查特征仍具有重要意义。

表1 不同临床类型WD患者发病年龄的特点Table 1 Age characteristics of different clinical types of Wilson’s disease
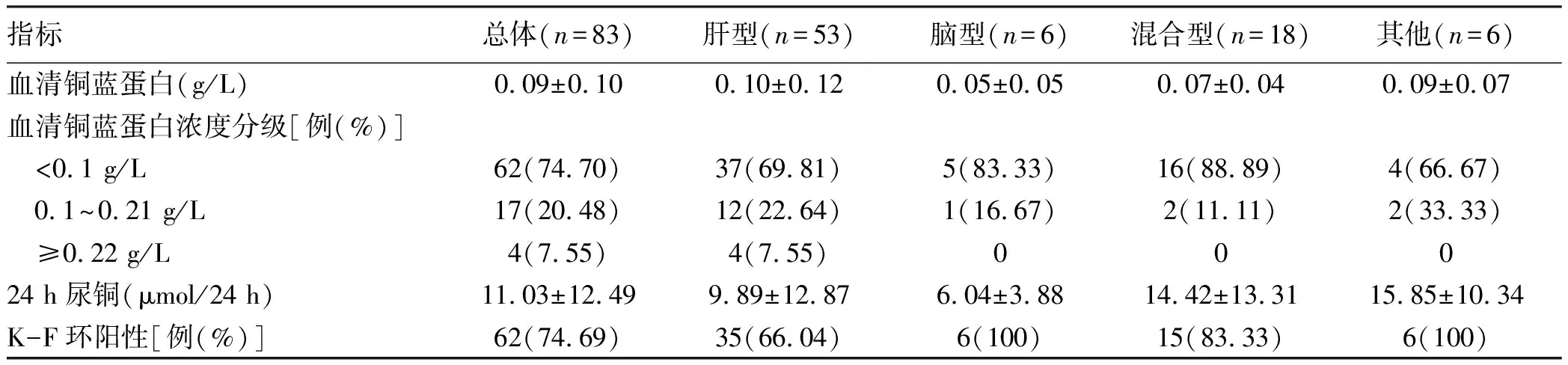
表2 不同临床类型WD患者铜代谢及角膜K-F环特征Table 2 Characteristics of copper metabolism and corneal K-F ring in different clinical types of Wilson’ s disease
本研究显示,WD患者年龄最小为3岁,其中年龄≤18岁者39例(46.99%),平均年龄(21.16±14.87)岁,首次发病年龄3~63岁,与既往文献[4]报道相符。我国曾有报道[5]肝酶水平升高、铜蓝蛋白水平下降的8月龄婴儿经基因检测确诊为WD,因此年龄不能作为排除本病的依据,相反婴幼儿及老年人出现不明原因肝损伤应警惕WD的可能。WD为常染色体隐性遗传病,既往报道[6]男性患者患病率略高于女性,其机制可能与雌激素水平与铁代谢差异有关,但本研究样本男女比例为1∶1.075,女性稍多于男性。WD有一定家族聚集倾向[7],本研究中5例患者因有明确家族史而早期诊断。因此,对已确诊WD患者的直系亲属应早期行ATP7B基因检测以明确诊断[8]。
本研究中,WD患者以肝病症状多见,临床分型以肝型和同时具有肝脏和神经系统表现的混合型为主,临床表现为急性肝炎、慢性肝炎反复发作、肝硬化,甚至肝衰竭。其中,已进展为肝硬化者31例(8例合并食管胃底静脉曲张出血,3例合并肝性脑病,5例因急性或慢加急性肝衰竭就诊),提示WD所致肝损伤起病隐匿,且发展迅速,若不能早期诊断和及时治疗,可发展致肝硬化甚至肝衰竭等不可逆的肝损伤,从而影响生存预后[9-10]。另有部分患者因查体发现肝脾肿大或肝功能异常就诊,无明显临床症状,如果接诊医师对WD缺乏足够认识,则易误诊为其他肝病[11]。本研究中,WD患者从发现异常到确诊WD历经1~37年,表明该病极易误诊或漏诊,应引起临床医师的高度重视。
WD脑型患者主要表现为震颤、口下颌肌张力障碍、运动迟缓及精神症状,与既往文献报道[12-14]大致相符,其中精神症状主要表现为记忆力下降、注意力不集中及学习成绩下降。本研究中,WD患者精神症状发生率为6.02%,低于既往报道[15-16]的14%~24.8%,考虑与精神症状早期表现不明显有关。WD的临床表现复杂多样,除常见的肝脏及椎体外系受损等,还可能出现骨骼系统受累[17]。本研究中,6例患者出现骨关节疼痛,确诊前一直误诊为骨关节疾病,这类患者往往因误诊为骨关节疾病而行手术治疗[18],应引起临床医师的注意。
血清铜蓝蛋白是诊断WD常用的实验室指标。本研究中,79例(95.18%)WD患者出现血铜蓝蛋白水平下降。然而,血清铜蓝蛋白并非WD的特异性指标,在营养不良、自身免疫性疾病患者中亦可见铜蓝蛋白水平下降,在重症感染、结核、胆汁淤积等情况下升高,具有一定的假阳性和假阴性可能。当过量的铜无法与铜蓝蛋白结合,也无法经胆道排出时,可出现尿铜水平升高。因此,24 h尿铜检测是诊断WD的有效手段。本研究中,73例(87.95%)WD患者尿铜水平升高。此外,K-F环也是WD的诊断标准之一。本研究中,K-F环阳性检出率仅为74.69%,与既往文献[19]报道较为一致。但K-F环阳性并非WD的特征性表现,亦可见于部分胆汁淤积性疾病中。有研究[20]显示,WD病程越长,就诊年龄越大,K-F分级越严重,而年龄较小的儿童患者的K-F环表现可能不明显。总之,上述这些单一指标诊断WD均有一定的局限性,参照Leipzig评分[21]行多指标联合检测并计算积分,积分≥4对确诊WD有明显帮助。
除了肝脏和神经系统等损伤以外,WD患者也可出现肾损伤,其机制可能与过量的铜离子沉积于肾小球和肾小管,影响肾脏的滤过和重吸收功能有关。本研究中,2例WD患者出现血肌酐水平升高,提示WD患者还应注意肾功能的检测。头颅CT/MRI可清晰显示颅脑结构及损伤,本研究中,17例WD患者行头颅CT或MRI检查,表现为基底节区(豆状核、尾状核、丘脑)对称性异常病灶,其次易受累的部位为脑干、大脑脚及脑白质变性,还可表现为脑萎缩,脑沟、池、裂增宽等,与既往文献[22]报道基本相符,但上述影像学表现应注意与其他神经系统疾病相鉴别。
近年来,ATP7B基因变异检测在WD诊断中的应用越来越广泛,基因检测阳性率为60%~85%[4],不同地区常见的ATP7B基因流行学特征及突变位点不同,亚洲人群的突变位点多为c.2333G>T/p.R778L,基因频率为34%~38%[23]。本研究样本的常见位点与既往报道相符,等位基因频率为60%,高于既往报道的频率。WD诊断是由临床到基因检测综合考虑确诊,基因检测未发现突变,不能排除本病。肝组织病理学检查是评估WD肝损伤及诊断的重要手段之一,但其肝脏形态学缺乏特征性改变,出现小泡性脂肪变性及铜沉积对诊断WD有一定提示作用[24]。本研究中,25例患者行肝组织病理学检查,其中10例患者为肝硬化阶段,12例为慢性肝炎阶段,这些患者肝组织铜沉积均较为明显;3例以肝细胞脂变为主,伴有少量铜颗粒沉积,应注意与其他原因引起的肝脂肪变鉴别。
综上所述,WD在各年龄均可发病,以青少年较多见,临床表现复杂多样且诊断困难,长期误诊或漏诊导致患者病情加重甚至死亡。因此,临床上如遇不明原因肝功能异常、肝硬化甚至肝衰竭患者,不明原因神经精神疾病患者,应注意筛查WD。血铜蓝蛋白、24 h尿铜、角膜K-F环可作为常规筛查指标,Leipzig评分可以提高诊断效能。诊断不清时可行肝组织病理学检查、头颅影像学检查或ATP7B基因检测,多指标联合分析,以提高临床诊断率,减少误诊和漏诊,改善患者预后。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:孔丽对提出研究思路、设计研究方案有关键贡献并负责文章最后修订;纪雷负责采集、分析数据并负责文章起草及修正。
猜你喜欢
中国现代医生(2022年21期)2022-08-22
医学概论(2022年3期)2022-04-24
中国典型病例大全(2022年7期)2022-04-22
广东教育·职教版(2021年3期)2021-04-20
中国药学药品知识仓库(2021年18期)2021-02-28
环球时报(2019-04-03)2019-04-03
学校教育研究(2018年27期)2018-05-14
小学科学(2016年10期)2016-09-10
科学中国人(2016年1期)2016-01-13
中国民族民间医药·下半月(2011年10期)2011-12-27
