
10例儿童肝糖原累积病临床及病理学分析
2022-09-08 03:09赵素贤刘世恒李文聪刘树红张庆山任伟光孔令波王荣琦南月敏赵景民
临床肝胆病杂志 2022年8期
赵素贤, 刘世恒, 李文聪, 韩 芳, 刘树红, 张庆山, 任伟光, 孔令波,付 娜, 王荣琦, 孔 丽, 南月敏, 赵景民
1 河北医科大学第三医院 中西医结合肝病科, 河北省肝纤维化机制研究重点实验, 石家庄 050051;2 解放军总医院第五医学中心 病理科, 北京 100039
肝糖原累积病(glycogen storage disease,GSD) 为先天性糖代谢酶缺陷所造成的糖原代谢障碍疾病, 多数属常染色体隐性遗传。至少有19型(包括亚型)[1],可分为影响肝脏或肌肉或两种组织的类型,由于酶缺乏的类型不同而表现出不同的临床症状、体征。临床表现缺乏特异性,易造成误诊,肝活检病理有助于本病的诊断[1]。现回顾性分析经肝穿刺活检病理证实的GSD 10例,作以临床分析。
1 资料与方法
1.1 研究对象 选择2002年1月—2022年1月河北医科大学第三医院及解放军第五医学中心通过临床及病理确诊的10例儿童GSD患者。
1.2 肝脏生化及免疫学指标检测 Olympus AU5400全自动生物化学分析仪检测ALT、AST、ALP、GGT、TBil、DBil、Alb及Glo;酶联免疫吸附试验检测抗HAV-IgM、HBsAg、抗-HBs、抗-HBc、HBeAg、抗-HBe、抗-HCV、抗-HDV、抗HEV-IgM及抗HEV-IgG;免疫荧光法检测抗核抗体、抗肝肾微粒体抗体、抗线粒体抗体及分型。
1.3 肝组织活检病理学检查 肝活检组织经4%甲醛液固定,石蜡包埋,连续5 μm组织切片,常规行HE、Masson染色,显微镜下观察炎症、纤维化程度及病变特点;普鲁士蓝染色及红氨酸染色观察肝细胞内铁及铜沉积情况。链霉亲和素-生物素复合物法检测肝组织原位HBsAg、HBcAg表达情况;PAS染色法观察肝组织内糖原沉积。
2 结果
2.1 一般资料 经临床病理诊断的GSD 10例,其中男7例,女3例,年龄2~9岁,症状出现最早者1岁时发现腹部膨大,余均为转氨酶升高原因待查入院。患儿均为足月产儿,其父母无近亲婚配,无家族遗传史,智力正常。
2.2 临床表现 本组患儿为缓慢起病,发育迟缓,矮小,均表现为肝功能异常,轻度乏力、纳差、尿黄、眼黄,4例患者肝脾肿大,其余患者肝脾肋下均未触及。6例患者有低血糖的临床表现(表1);1例患儿经常摔倒,双侧腓肠肌肥大,Gower征阳性。
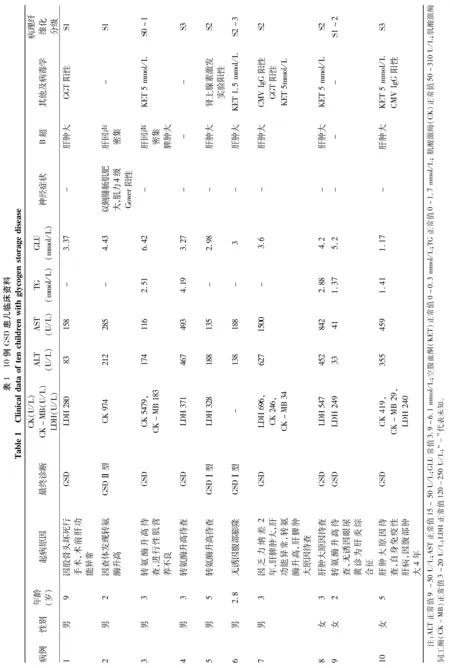
2.3 实验室检查 6例空腹血糖为低血糖,血糖正常者3例,有1例血糖偏高。肝功能:9例AST波动在116~842 U/L,9例ALT波动在138~627 U/L,4例出现肌酶谱升高。TC均正常,5例TG升高;5例尿酮体升高;10例患者甲-戊型肝炎标志物均为阴性,铜蓝蛋白均正常。10例自身抗体、角膜色素环均为阴性,2例患儿巨细胞病毒IgG阳性。B超提示肝肿大6例,脾肿大1例,其中3例行基因检测,2例为GSDⅠ型,1例为GSDⅡ型(表1)。
2.4 病理学特点 10例患者均经肝穿刺活检病理确诊。其中10例肝组织镜下见肝细胞弥漫性肿大,胞浆空淡,核小居中似植物细胞状(图1),汇管区炎性细胞浸润,5例间质纤维组织轻度增生,纤维间隔形成;其中2例小叶结构紊乱,10例肝糖原染色(PAS法)均为阳性(图2)。病理诊断为GSD。

图1 HE染色结果(×200)Figure 1 HE staining(×200)

图2 PAS染色结果(×200)Figure 2 Periodic Acid-Shiff’s staining(×200)
2.5 治疗及随访 保肝治疗基础上,进行营养支持治疗。饮食上控制甜食的摄入,少量多餐,高碳水化合物、足量蛋白质、低脂饮食,并给予生玉米淀粉口服治疗;每次0.6~2.0 g/kg,1次/6 h。经治疗患儿肝功能恢复正常,肝肿大明显好转。随访2年时,患者肝肿大明显好转,无低血糖发生,病例1身高未见明显变化,其余患者身高均不低于同龄患者平均身高。
3 讨论
GSD是一种少见的隐性遗传性疾病,是一类由于先天性酶缺陷所造成的糖原代谢障碍疾病。其特点为糖合成和分解代谢的酶缺陷致糖中间代谢紊乱。当其中某一个或几个酶缺陷时,主要生化反应途径受阻,可有正常结构或不典型结构的糖原累积于肝、肾、骨骼肌、心肌及中枢神经系统等组织,结果出现肝脾肿大、肌张力降低或肌痉挛、低血糖、乳酸血症等临床表现。
本研究患者均表现出身材矮小,多数有肝肿大,肝功能异常,可能由于糖原累积于肝脏,导致肝肿大,肝功能不全,蛋白质代谢紊乱,体格矮小。本研究结果表明患儿有肝功能异常表现,部分有TG升高及低血糖,但也有例外,故临床表现各异。但这类疾病有一个共同的生化特征,即是糖原贮存异常,依据组织酶学检查结果,其中累及肝脏者最多为Ⅰ型,其次为Ⅱ、Ⅲ、Ⅳ、Ⅵ型[2],本研究中仅3例患者行基因检测,结果回报为Ⅰ型和Ⅱ型,临床表现为转氨酶异常,因此肝功能异常患儿就诊时,应注意糖原累积病,具体分型需依赖基因检测。
Ⅰ型GSD又称Von Gierke 病,有家族性遗传倾向,近亲结婚者易于发生。实验室检查常见空腹低血糖、高脂血症和乳酸增高。Ⅱ型GSD,亦称Pompe病,本病血糖、血脂正常,无酸中毒,对胰高血糖素和肾上腺素试验反应正常。Ⅲ型GSD(Forbes病)系缺乏淀粉-1,6-葡萄糖苷酶(脱支链酶)所致,病变主要累及肝、肌肉和心脏。临床表现与生化检查与Ⅰ型相似,但一般较轻。肝肿大、肌肉容易疲劳,早年生长发育延缓,随年龄增长而好转,有的可发展为肝硬化。实验室检查有低血糖、高脂血症,但乳酸不增高。
本组部分患儿肝组织穿刺表现为肝组织结构紊乱,Ⅳ型糖原累积病(Aderson病)病理学上为小结节性肝硬化,肝、脾、淋巴结、肠黏膜有少量糖原沉积、单核巨噬细胞系统有显著的支链淀粉样糖原颗粒。临床以肝硬化、腹水、出血及肌无力为主要表现,并有中度低血糖,多死于肝衰竭。因此患儿临床提示严重肝纤维化时,注意排查Ⅳ型GSD。
因各型GSD临床表现和血生化检查不一,本病需要与其他儿童疾病相鉴别,鉴别诊断总结如下。(1)肝豆状核变性[3-4]:由于本病多见于儿童,是一种常染色体隐性遗传性铜累积病,该病一般都有家族聚集性倾向,主要表现为因铜蓝蛋白缺乏,铜结合不足或铜排入毛细胆管障碍造成过量的铜沉积于肝脏、脑核、虹膜等处,儿童时期可表现为肝病症状,肝活检组织铜染色结合血清铜蓝蛋白及基因检测可确诊本病。(2)病毒性肝炎:因多数患者起病隐匿,表现为查体发现肝功能异常或表现为乏力、纳差、尿黄、眼黄;故需要排除嗜肝及非嗜肝病毒感染。(3)癫痫[5]:低血糖可致婴儿反复发生惊厥、抽搐、昏迷、智能减退, 严重者可发生酮症酸中毒和继发感染临床常因反复抽搐误诊为“癫痫”。(4)多发性肌炎[6]: Ⅱ型GSD多在儿童和青春期发病,可表现为在较强的体力活动后很快感到疲劳,骨骼肌在短时间激烈活动后出现疼痛、痉挛、无力,休息后可缓解,部分患者可在后期出现持久的肌萎缩和肌无力。而多发性肌炎是以肌痛、近端肌无力为主要表现的肌肉炎性肌病,实验室检查有肌酶谱增高,红细胞沉降率加快,肌电图表现为肌源性损害。(5)肝转移肿瘤[7]:本病B超可表现为肝脏外形增大, 表面欠光滑, 肝实质回声分布不均匀, 肝内可见多个混合性回声团块。有研究[8]表明, GSD的肝脏超声表现为不同程度的脂肪肝, 包括均匀性脂肪肝及非均匀性脂肪肝,非均匀性脂肪肝中的局灶性脂肪肝应与肝肿瘤相鉴别, 结合病史及相关的实验室检查, 应考虑到GSD的可能, 鉴别困难时可行超声引导下穿刺活检确定诊断。
综上所述,本组患者起病隐袭,多以肝功能异常或肝肿大为首诊原因,但因本病临床分型较多,临床表现各异,曾有各种误诊报道如上鉴别诊断中的病毒性肝炎、癫痫、多发性肌炎等,故对于儿童及青少年有相关临床表现的病人,肝炎病毒标志阴性,无其他原因可解释者,应考虑GSD,进一步行肝穿刺活检及基因测序分析明确诊断及肝组织病变程度[9-10]。
伦理学审查:本研究方案于2019年12月25日经由河北医科大学第三医院伦理委员会审批,批号:K2019-014-2。所纳入患者均由其监护人签署知情同意书。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突,特此声明。
作者贡献声明:赵素贤、刘世恒负责收集数据,撰写论文;李文聪、韩芳负责资料分析;张庆山、任伟光、孔令波参与临床病例收集;付娜、王荣琦、孔丽进行文献检索及分析;赵素贤、刘树红参与病理解读并提供病理图片;赵景民参与课题设计;南月敏参与课题设计,拟定写作思路,指导撰写文章并最后定稿。
猜你喜欢
西北民族大学学报(自然科学版)(2022年2期)2022-07-06
肝博士(2022年3期)2022-06-30
家庭医学(2020年11期)2020-12-28
中国医药导报(2019年3期)2019-03-18
商情(2018年9期)2018-03-29
考试周刊(2018年8期)2018-01-19
婚育与健康(2017年9期)2017-11-04
中国市场(2017年5期)2017-03-15
现代养生·下半月(2016年6期)2016-10-21
求医问药(2009年7期)2009-08-31
