
高效红光铱配合物的合成、光物理性质和理论计算
2022-09-21 12:03葛泽荣赵世盛李文豪李红岩
云南大学学报(自然科学版) 2022年5期
佟 鑫,葛泽荣,赵世盛,李文豪,楚 曦,李红岩
(河北工业大学 化工学院,天津 300130)
在过去的几十年中,铱配合物作为一种磷光发光材料因其发光效率高、激发态寿命相对较短、斯托克斯位移较大以及发光颜色可调节等优点[1-4]迅速发展起来并被应用于有机发光二极管(OLEDs)、细胞成像和化学传感等不同领域[5-9].金属铱能使配合物产生强自旋轨道耦合作用,并且铱金属离子中的 d 轨道分裂较大,可尽量减弱与配合物的金属−配体电荷转移跃迁(MLCT)态相互作用,这对提升磷光发光效率有积极作用.铱的3 价离子可与配体形成非常稳定的中性和离子型分子.中性铱配合物已广泛应用于有机电致发光器件中,获得了高性能的红、绿、蓝等其他各种发光颜色的OLEDs器件[10].相较于中性铱配合物来说,阳离子铱配合物的研究稍显缓慢.阳离子铱配合物,除了具有中性铱配合物高的发光效率、颜色可调等特点外,还具有溶解性好,易合成提纯等优点[11].目前被广泛研究的是以 [Ir(C^N)2(N^N)]PF6形式为主的阳离子铱配合物,如何通过结构设计,利用此类型配合物获得高效红光发光材料是近期的研究热点.为了使铱配合物的发光红移,一般采用调整分子结构的方法来减小最低空轨道(LUMO)和最高占据轨道(HOMO)间的能隙[12].根据文献[13-14],实现铱配合物发光红移的常用方法可归纳为以下3 点:①在环金属配体的苯基上引入给电子基团;② 将苯并喹啉基团上的C 原子置换成N 原子;③引入亚甲基或稠环结构扩大π 共轭体系.由此看出,通过环金属配体和中性配体的分子设计,调控配合物的分子轨道能级,可以实现铱配合物的高效红光发射.
在本文中,我们合成了基于4,4′−二叔丁基−2,2′−联吡啶(dtb-bpy)为中性配体,1−(3−氟苯基)异喹啉(3F−piq)为环金属配体的离子型铱配合物[(3F−piq)2Ir(dtb-bpy)](PF6)(Ir1).选择1−(3−氟苯基)异喹啉为环金属配体,通过扩大其π 共轭体系,使配合物的发射光谱红移,获得性能优良的红光材料.详细研究了[(3F−piq)2Ir(dtb-bpy)](PF6)的晶体结构、光物理性质、电化学性质,并对配合物的前线分子轨道的电子密度分布以及电子跃迁情况进行了分析[15].
1 实验部分
1.1 试剂和仪器1−氯异喹啉(分析纯,北京百灵威科技有限公司)、3−氟苯硼酸(分析纯,天津希恩斯生化科技有限公司)、四(三苯基膦)钯(分析纯,陕西开达化工有限公司)、IrCl3·3H2O(昆明铂锐金属材料有限公司)、KPF6(分析纯,阿拉丁试剂有限公司)、4 4′−二叔丁基−2,2′−联吡啶(dtb-bpy,分析纯,阿拉丁试剂有限公司)、2−乙氧基乙醇(分析纯,北京百灵威科技有限公司).
核磁共振氢谱采用Bruker AM 400 MHz 核磁共振仪完成;晶体由Siemens(Bruker)SMART CCD单晶衍射仪测定;用密度泛函(DFT)理论,B3LYP方法对配合物进行了几何结构优化,Ir 金属原子采用有效赝势(ECP),LANL2DZ 方法,其他原子则使用6−31G(d,p)基组,所有的计算都是通过Gaussian09程序包完成;循环伏安曲线测量在CHI 760E 电化学工作站进行;吸收光谱和发射光谱分别在UV−2700 紫外−可见分光光度计和Hitachi F−2700 荧光光谱仪上测得;磷光寿命是在Edinburgh FLS920P光谱仪上通过时间分辨实验在MLCT 激发和最大发射波长处测得,并经过拟合处理得到;发光量子效率是以除氧的Ir(ppy)3溶液(Φstd=0.97)为标准物质,根据文献[16]计算得到.
1.2 配合物的合成合成路线如图1 所示.环金属配体1−(3−氟苯基)异喹啉(3F−piq)按照文献[17]报道的方法进行合成.
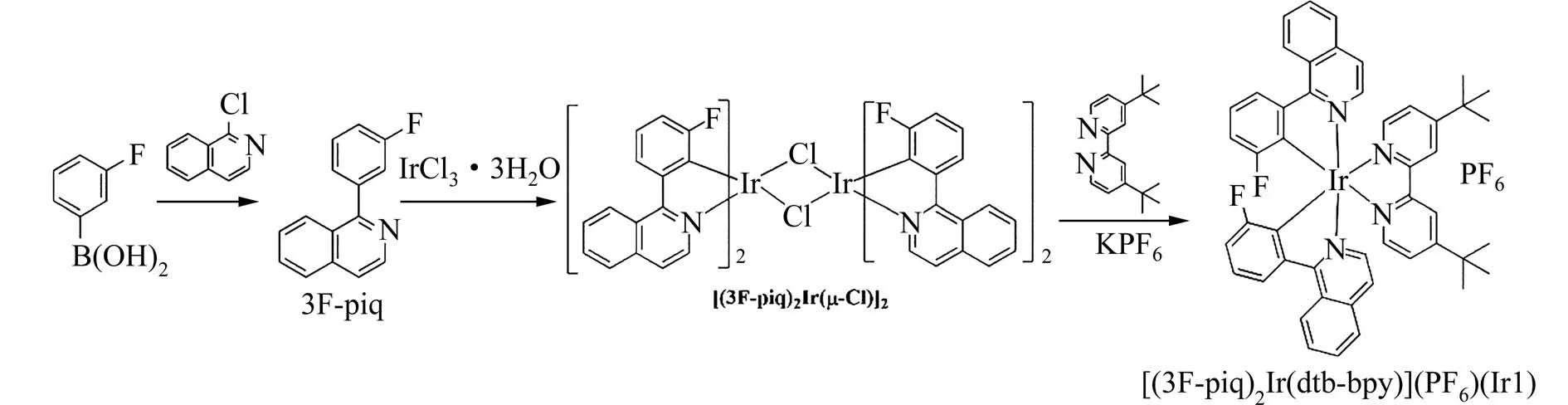
图1 配合物Ir1 的合成路线Fig.1 Synthetic routes of the complex Ir1
1.2.1 铱二氯桥配合物[(3F−piq)2Ir(μ−Cl)]2的合成在氮气保护下,将0.35 g(1.0 mmol)IrCl3·3H2O 和0.56 g(2.5 mmol)3F−piq 溶解在12 mL的体积比为3∶1 的2−乙氧基乙醇和水的混合溶剂中,140 ℃下反应10 h.冷却后,有黄色[(3F−piq)2Ir(μ−Cl)]2沉淀析出,过滤,分别用乙醇和水洗涤.产物在真空下干燥,得到0.60 g 铱二氯桥配合物[(3F−piq)2Ir(μ−Cl)]2,产率90%.
1.2.2 配合物[(3F−piq)2Ir(dtb-bpy)](PF6) (Ir1)的合成在氮气保护下,将0.54 g(0.40 mmol)[(3F−piq)2Ir(μ−Cl)]2和0.27 g(1.0 mmol)的4,4′−二叔丁基−2,2′−联吡啶溶解在30 mL 二氯甲烷和甲醇的混合溶剂(体积比为1∶1)中,加热回流反应10 h,冷至室温后,加入0.36 g(2.0 mmol)KPF6固体,室温反应2 h.反应结束后减压蒸干溶剂,用二氯甲烷−石油醚(体积比1∶1)做扩展剂进行硅胶柱层析,得到0.28 g 橙红色的配合物Ir1,产率33%.1H NMR(400 MHz,DMSO−d6)δ:9.05~8.87(m,4H),8.33~8.23(m,1H),8.18(d,J=11.7 Hz,1H),8.11(s,1H),8.04(d,J=4.6 Hz,1H),7.98~7.81(m,4H),7.73~7.49(m,7H),7.45~7.35(m,1H),7.28~7.13(m,1H),6.89(d,J=8.5 Hz,1H),6.68(dd,J=10.5,6.3 Hz,1H),6.13~5.96(m,1H),1.46~1.28(m,18H).MS(ESI),m/z:905.30 [M−PF6]+.
2 结果与讨论
2.1 晶体结构室温下,配合物Ir1 的单晶是通过CH2Cl2和CH3OH 的混合溶液自然挥发得到的.配合物Ir1 的晶体结构如图2 所示.晶体结构分析表明Ir1 属于单斜晶系,P2(1)空间群.对应晶体学参数见附件表S1,部分键长键角列于附件表S2.图2 中Ir 中心为六配位结构,其几何构型均为扭曲的八面体构型,其八面体由环金属配体3F−piq 的2 个C 原子和2 个N 原子,和中性配体的2 个N原子构成.在配合物Ir1 中,Ir—C 键键长处于0.201 1(5)~0.201 6(5)nm 范围内,Ir—N 键键长处于0.204 6(5)~0.212 7(4)nm 范围内,配合物中其他键长键角也都处于正常范围之内.从表S2 中可以看出,金属Ir 与中性配体形成的Ir—N 配位键(Ir1—N3 和Ir1—N4)的键长比Ir 与环金属配体形成的Ir—N键(Ir1—N1 和Ir1—N2)略长一些,说明中心金属与不同类型的配体形成的配位键有差别,与环金属配体形成的Ir—N 键更牢固.对于配合物Ir1,中性配体4,4′−二叔丁基−2,2′−联吡啶和环金属配体之间的二面角接近90°,分别为89.1°和89.2°.

图2 配合物Ir1(CCDC:1884253)的晶体结构Fig.2 Crystal structure of complex Ir1(CCDC: 1884253)
2.2 光物理性质图3(a)所示是配合物Ir1 在室温条件下、二氯甲烷溶液(2×10−5mol/L)中的紫外−可见吸收光谱图,吸收峰和摩尔吸收系数等数据列于表1 中.从图3(a)中可以看出,配合物Ir1 在230~320 nm 范围内有较强的吸收峰,摩尔吸收系数17~64 kmol−1·L·cm−1,这些吸收可以归属于以配体为中心的自旋允许的π→π*的跃迁,由于是电子自旋允许,所以有着较高的摩尔吸光系数.在320~400 nm范围内,配合物Ir1的吸收峰位于355 nm.这个吸收带主要由配体到配体之间的电荷转移跃迁(LLCT)和配体内的电荷转移跃迁(ILCT)吸收产生.在能量较低的400~550 nm 范围内的吸收可以归属为金属−配体间的电荷转移跃迁吸收,即MLCT 吸收,包括自旋允许的1MLCT 和自旋禁阻的3MLCT 的混合跃迁吸收.
图3(b)所示是配合物Ir1 在二氯甲烷溶液(2×10−5mol/L)中的发射光谱.配合物Ir1 的最大发射波长、量子效率和寿命等数据汇总于表1 中.在327 nm 的激发条件下,配合物Ir1 在二氯甲烷溶液中的发射峰为590 nm,色坐标为(0.59,0.41),溶液呈现明亮的红光.以标准物质Ir(ppy)3为参比,根据配合物在除氧的二氯甲烷中的发射光谱和吸收光谱计算得到配合物Ir1 的量子效率为55%.配合物Ir1 在除氧的二氯甲烷溶液中的磷光寿命为2.33 μs.这些光物理性质表明,配合物Ir1 为高效的红色磷光发光材料.
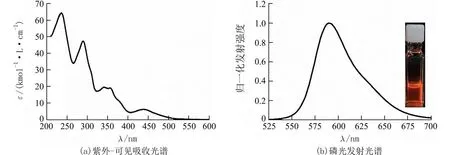
图3 配合物Ir1 在二氯甲烷溶液中的光谱Fig.3 The spectra of complex Ir1 in dichloromethane solution
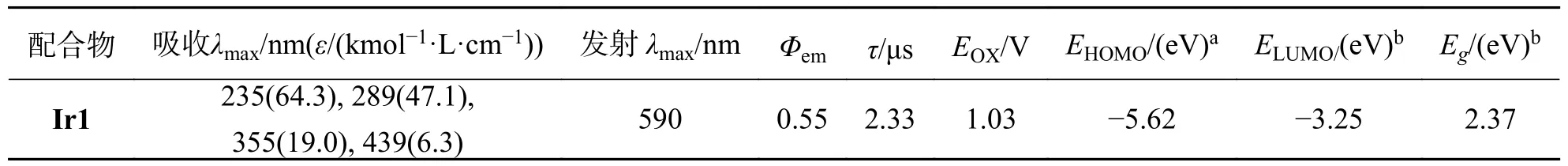
表1 配合物Ir1 的光物理性质和电化学性质相关数据Tab.1 Photophysical and electrochemical data for complexes Ir1
2.3 电化学性质配合物Ir1 在CH3CN/CH2Cl2(体积比1∶1)溶液中的循环伏安曲线如图4 所示,相关电化学数据见表1.配合物Ir1 在0.8~1.5 V范围内出现一组可逆的氧化峰,这一过程归因于Ir3+/Ir4+的氧化还原过程.以二茂铁的费米能级4.8 eV 为参照,根据配合物的氧化电位计算配合物的最高占据分子轨道(HOMO)能级为−5.62 eV.根据配合物Ir1 的紫外−可见吸收光谱和发射光谱,得到HOMO和最低空分子轨道(LUMO)的能隙差Eg,再进一步计算得到LUMO 轨道能级为−3.25 eV.配合物的氧化还原性质与HOMO、LUMO 能级及其能隙直接影响该物质的载流子传输性能和发光颜色,并且对器件结构的设计也非常关键[18-19].

图4 配合物Ir1 的循环伏安曲线Fig.4 The cyclic voltammogram for complex Ir1
2.4 理论计算为探究铱配合物的前线分子轨道的电子分布、能级以及电子跃迁性质,采用Gaussian09程序对配合物Ir1 进行了理论计算.配合物Ir1 的HOMO−1、HOMO、LUMO 和LUMO+1轨道能级及电子密度分布如图5 所示,各轨道电子密度百分比构成列于附件表S3 中.图6 为理论计算得到的紫外−可见吸收光谱与实验光谱对比图.附件表S4为通过含时密度泛函方法(TD−DFT)计算得到的主要电子跃迁位置和性质,TD−DFT 计算对于准确地指认紫外−可见吸收光谱的归属是一个非常好的手段.
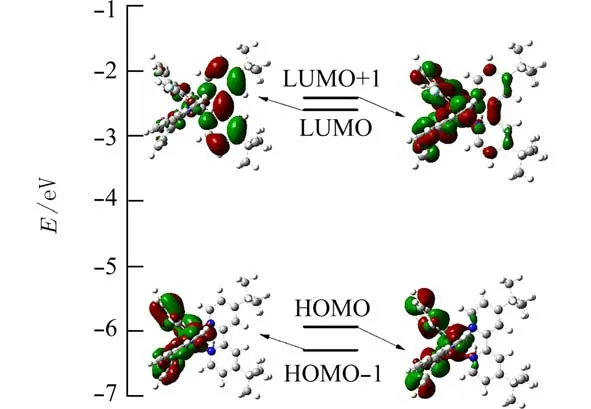
图5 DFT 理论计算得到的配合物Ir1 的前线分子轨道能级图Fig.5 The frontier molecular orbital diagrams of the complex Ir1 by DFT calculations

图6 理论计算得到的紫外−可见吸收光谱与实验光谱对比图Fig.6 The comparison between the theoretically calculated UV-vis absorption spectrum and the experimental spectrum
从图5 和附件表S3 可以看出,配合物Ir1 的HOMO 轨道主要由金属铱(45.19%)和环金属配体(52.18%)组成,HOMO−1 轨道中金属铱的成分减少为21.84%,环金属配体增加至76.50%.而LUMO轨道主要分布在中性配体(84.89%)上,LUMO+1轨道主要分布在环金属配体(81.36%)上.理论计算结果对于配合物的结构修饰具有一定的指导意义.对于中性配体的修饰,主要影响LUMO 轨道能级,在中性配体上引入吸电子基团会稳定LUMO 轨道能级.而对于环金属配体的修饰,主要影响HOMO轨道能级,在环金属配体上引入供电子基团或增大共轭体系,会使HOMO 轨道能级升高,从而减少HOMO 和LUMO 轨道的能隙差,使光谱红移.表明TD−DFT 理论计算模拟得到的紫外−可见吸收光谱图和实验谱图的吻合度较好.从图6 表S4可以看出,高能量区域的电子跃迁主要归属于LLCT 和ILCT 过程,而低能量区域的电子跃迁主要归属于MLCT 跃迁,理论计算结果能够很好地解释由实验得到的紫外−可见吸收光谱的电子跃迁性质.
3 结论
以1−(3−氟苯基)异喹啉为环金属配体,以4,4′−二叔丁基−2,2′−联吡啶为中性配体,六氟磷酸根为阴离子合成了一种新型的离子型铱配合物Ir1,并进行了结构表征、理化性质和理论计算研究.在二氯甲烷溶液中,配合物Ir1 在230~550 nm 范围内具有较强的吸收,发射峰位于590 nm,溶液呈现明亮的红光,量子效率较高为55%.理论计算表明,在环金属配体上引入供电子基团或增大共轭体系,会使HOMO 轨道能级升高,从而减少HOMO 和LUMO 轨道的能隙差,使光谱红移.理论计算结果对于该类型配合物的结构修饰以及得到高效磷光发光材料具有一定的指导意义.
猜你喜欢
中国防痨杂志(2022年7期)2022-11-25
昆钢科技(2022年2期)2022-07-08
昆钢科技(2022年2期)2022-07-08
中国防痨杂志(2022年3期)2022-03-11
昆钢科技(2021年4期)2021-11-06
科技与创新(2020年16期)2020-08-18
世界家苑(2018年9期)2018-09-18
分析化学(2018年12期)2018-01-22
科技与创新(2015年20期)2015-10-29
科技与企业(2015年20期)2015-10-21
