
马来酸氟伏沙明-D3的合成
2022-10-11 14:03仇中选黄龙江
合成化学 2022年9期
仇中选, 王 东, 黄龙江
(1. 青岛职业技术学院 生物与化工学院,山东 青岛 266555; 2. 青岛科技大学 化工学院,山东 青岛 266042)
马来酸氟伏沙明是由雅培制药研发的一种治疗抑郁症和强迫症药物,是全球第一种选择性5-羟色胺再摄取抑制剂(SSRIs)类型的抗抑郁药物,也是唯一一种单环类SSRIs[1-4]。与传统的抗抑郁药物相比,马来酸氟伏沙明具有拟肾上腺素效能低,不易引起兴奋、多汗等症状;抗胆碱和抗组胺效能低,很少引发体重增加和嗜睡等现象;药物间相互作用小,有利于联合用药等优点。临床统计表明,马来酸氟伏沙明用药时对胃肠道、中枢神经系统、性功能等方面具有一定副作用[5-7]。
氘代标记化合物是利用氘的同位素效应,将化合物中的氢原子或部分氢原子用氘原子替换的化合物,在生物医学、药物代谢动力学中都发挥着重要作用[8-10]。美国食品药品监督管理局(FDA)颁布的工业指南要求在新药研究中,药物代谢安全评价必须使用同位素标记技术。将氘代技术应用到现有药物的合成中,不会影响分子构象,有效性与安全性经过验证,可用于改善早期开发药物的部分缺点及副作用。本研究将马来酸氟伏沙明中的甲基进行氘代化,合成稳定同位素标记的马来酸氟伏沙明-D3,有助于进一步分析马来酸氟伏沙明在人体内的代谢过程以及药物动力学,为改善其副作用奠定基础。
目前,国内稳定同位素标记药物的研发和生产处于起步阶段,市售的马来酸氟伏沙明-D3为代理进口产品,价格昂贵,关于其合成技术还未见文献报道,严重限制了该标准品在我国的广泛使用。已报道的马来酸氟伏沙明的合成方法主要有两种:(1)以对三氟甲基苯腈为原料,与格氏试剂反应直接制备目标化合物[11-14]。该路线是目前文献报道较多的方法,但反应周期长、重复性差,且使用高毒性氰类化合物为原料,使用时存在安全隐患。(2)以对三氟甲基苯甲酸为原料,在PCl3催化下,与甲氧基甲基胺反应生成Weinreb酰胺,进而与格氏试剂反应得到马来酸氟伏沙明[15]。该反应过程用到PCl3,易造成环境污染,产生大量酸性废液,增加了三废处理成本,且不适合甲基氘代化。因此,探索一种稳定性高、简洁高效合成马来酸氟伏沙明-D3的方法是一项具有现实意义的工作。
本文首次以市售的4-苄氧基-1-丁醇(2)为原料,与氘代碘甲烷反应,得到氘代甲基化中间体(3),后续经过氢化脱苄、溴化、格氏反应、戴斯-马丁氧化、缩合、取代、成盐等反应,合成高纯度马来酸氟伏沙明-D3(1, Scheme 1),具体合成路线如Scheme 1所示。该化合物的合成方法现已申请专利保护,公开号为CN 113861066[16]。
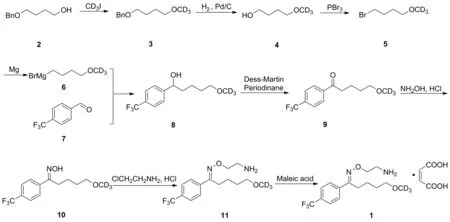
Scheme 1
1 实验部分
1.1 仪器与试剂
Bruker AC-500 MHz型核磁共振波谱仪;Ultima Global spectrometer型高分辨质谱仪(德国英福康公司);WATERS-e2695型高效液相色谱仪(美国沃特斯)。
4-苄氧基-1-丁醇、三溴化磷、盐酸羟胺、2-氯代乙基胺盐酸盐、马来酸、碘、4-三氟甲基苯甲醛(99%,阿达玛斯试剂有限公司);氘代碘甲烷(D: 99.9%,阿达玛斯试剂有限公司);氢化钠(60%,阿达玛斯试剂有限公司);无水四氢呋喃(Water≤50 ppm,99.5%,阿达玛斯试剂有限公司);Dess-Martin试剂(97%, Sigma-Aldrich);镁屑(99%, Sigma-Aldrich);钯碳催化剂(干基,10% Pd/C,阿拉丁试剂有限公司);其余所用试剂均为分析纯,青岛华东化学试剂有限公司。
1.2 合成
(1) ((4-(甲氧基-D3)丁氧基)甲基)苯(3)
500 mL三口瓶中,将4-苄氧基-1-丁醇(1, 18.0 g, 100 mmol)溶于N,N-二甲基甲酰胺(DMF, 200 mL),体系降温至0~5 ℃,搅拌下加入氢化钠(60%,分散于液状石蜡)(6.0 g, 150 mmol),加毕,搅拌10 min;滴加氘代碘甲烷(29.0 g, 200 mmol),滴毕,反应体系自然升至室温,搅拌反应12 h。体系倒入冰水中,加入乙酸乙酯(200 mL),分液,有机相依次用水(200 mL)、饱和食盐水(200 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩得无色液体319.3 g,收率98%,不作纯化直接下一步投料。
(2) 4-(甲氧基-D3)丁-1-醇(4)
在500 mL三口瓶中,将((4-(甲氧基-D3)丁氧基)甲基)苯(3, 17.7 g, 90 mmol)溶于无水甲醇(180 mL),加入钯碳催化剂(1.77 g,钯碳催化剂中钯含量为10 wt%),于氢气气氛中室温下快速搅拌12 h。停止反应,过滤,滤饼用无水甲醇(10 mL)洗涤,滤液减压浓缩至恒重得4-(甲氧基-D3)丁-1-醇(4)9.5 g,收率98%,无色液体,不作纯化直接下一步投料。
(3) 1-溴-4-(甲氧基-D3)丁烷(5)
在500 mL三口瓶中,氩气保护,在大约0~5 ℃左右温度下,将三溴化磷(24.6 g, 91 mmol)缓慢滴加至4-(甲氧基-D3)丁-1-醇(4, 7.5 g, 70 mmol)的四氢呋喃(140 mL)溶液中,加毕,自然升至室温搅拌12 h。加入饱和碳酸氢钠溶液调pH至7~8,乙酸乙酯(150 mL)萃取分液,有机相依次用水(100 mL)、饱和食盐水(100 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,柱层析分离得1-溴-4-(甲氧基-D3)丁烷(5)9.9 g,收率83%,无色液体。1H NMR(400 MHz, CDCl3)δ: 3.53~3.41(m, 4H), 2.05~1.91(m, 2H), 1.81~1.68(m, 2H);13C NMR(125 MHz, CDCl3)δ: 72.7, 65.2, 34.1, 29.8, 28.3。
(4) (4-(甲氧基-D3)丁基)溴化镁(6)
氩气保护下,250 mL三口瓶中加入镁屑(1.44 g, 60 mmol),一小粒碘、无水四氢呋喃(30 mL),加热至微沸,体系棕色褪去,滴加1-溴-4-(甲氧基-D3)丁烷(5, 8.5 g, 50 mmol)的无水四氢呋喃(30 mL)溶液,保持微沸状态,加毕,搅拌1 h制得(4-(甲氧基-D3)丁基)溴化镁(6)的四氢呋喃溶液。
(5) 5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)
在氩气保护下,0~5 ℃左右的温度下,将4-三氟甲基苯甲醛(7, 8.7 g, 50 mmol)的无水四氢呋喃(20 mL)溶液滴加至上步制得的(4-(甲氧基-D3)丁基)溴化镁(6)的四氢呋喃溶液中,加毕,自然升至升温搅拌3 h,加入1 mol/L盐酸(10 mL)淬灭反应,加入水(100 mL)、乙酸乙酯(100 mL)萃取分液,有机相用饱和食盐水(100 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩,柱层析分离得5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)11.1 g,两步收率84%,白色固体;1H NMR(400 MHz, CDCl3)δ: 7.60(d,J=6.4 Hz, 2H), 7.46(d,J=6.4 Hz, 2H), 4.76(s, 1H), 3.36(t,J=5.1 Hz, 2H), 2.16(d,J=2.2 Hz, 1H), 1.81~1.71(m, 2H), 1.63(s, 1H), 1.62~1.58(m, 2H), 1.51~1.48(m, 1H), 1.41~1.36(m, 1H);13C NMR(125 MHz, CDCl3)δ: 147.8, 128.7, 125.1, 124.4, 124.3, 122.1, 72.7, 71,4, 37.9, 28.3, 21.3。
(6) 5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)
500 mL反应瓶中加入5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8, 10.6 g, 40 mmol)、Dess-Martin试剂(25.4 g, 60 mmol)和二氯甲烷(100 mL),室温搅拌2 h,加入饱和碳酸氢钠溶液(100 mL),搅拌10 min,萃取分液,水相用二氯甲烷(100 mL)萃取,合并有机相,用饱和食盐水(100 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩、干燥得5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)10.1 g,收率96%,白色固体。1H NMR(400 MHz, CDCl3)δ: 8.05(d,J=6.5 Hz, 2H), 7.72(d,J=6.5 Hz, 2H), 3.42(t,J=5.1 Hz, 2H), 3.02(t,J=5.6 Hz, 2H), 1.85~1.80(m, 2H), 1.69~1.67(m, 2H);13C NMR(125 MHz, CDCl3)δ: 198.8, 139.4, 134.8, 128.1, 125.4, 125.3, 124.4, 72.1, 63.8, 38.3, 28.8, 20.6。
(7) (E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10)
250 mL反应瓶中加入盐酸羟胺(3.1 g, 45 mmol)、氢氧化钾(5.1 g, 90 mmol)和无水乙醇(100 mL),室温搅拌30 min,加入5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9, 7.9 g, 30 mmol),回流3 h。停止反应,降至室温,加入水(100 mL)、二氯甲烷(100 mL)萃取分液,水相用二氯甲烷(50 mL)萃取,合并有机相,用饱和食盐水(100 mL)洗涤,无水硫酸钠干燥,抽滤,滤液减压浓缩、柱层析得(E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10)7.8 g,收率91%,白色固体。1H NMR(400 MHz, CDCl3)δ: 7.71(d,J=6.5 Hz, 2H), 7.53(d,J=6.5 Hz, 2H), 3.40(t,J=5.0 Hz, 2H), 2.68(t,J=5.6 Hz,2H), 1.75~1.67(m, 2H), 1.53~1.47(m, 2H);13C NMR(125 MHz, CDCl3)δ: 157.5, 134.1, 133.3, 127.6, 125.1, 124.9, 124.4, 72.3, 63.3, 36.9, 28.0, 19.9。
(8) (E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11)
250 mL反应瓶中加入(E)-5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊酮肟(10, 7.0 g, 25 mmol)、2-氯代乙基胺盐酸盐(2.9 g, 25 mmol)、氢氧化钾(3.1 g, 55 mmol)和N,N-二甲基甲酰胺(80 mL),室温搅拌6 h。停止反应,减压浓缩,残渣加入2 mol/L盐酸调pH大约至1~2,加入水(100 mL)、二氯甲烷(100 mL)萃取分液,水相用10%氢氧化钠水溶液调pH大约至11~13,用二氯甲烷(100 mL)萃取,有机相用无水硫酸钠干燥,抽滤,滤液减压浓缩至恒重得(E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11)7.8 g,收率92%,无色液体,不作纯化直接下一步投料。
(9) 马来酸氟伏沙明-D3(1)
250 mL反应瓶中加入(E)-5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮-O-(2-氨基乙基)肟(11, 6.4 g, 20 mmol)、马来酸(2.3 g, 20 mmol)和无水乙醇(60 mL),室温搅拌2 h,滤液减压浓缩得马来酸氟伏沙明-D3粗品,加入乙酸乙酯和正己烷的混合液(40 mL,其中乙酸乙酯和正己烷的体积比为1 ∶9),打浆1 h,抽滤,滤饼用正己烷(20 mL)洗涤,干燥得马来酸氟伏沙明-D3(1) 8.4 g,收率96%,纯度99.3%,白色固体。1H NMR(400 MHz, CDCl3)δ: 8.22(brs, 2H), 7.66(d,J=6.6 Hz, 2H), 7.56(d,J=6.6 Hz, 2H), 6.14(s, 2H), 4.39(t,J=3.5 Hz, 2H), 3.38~3.32(m, 4H), 2.73(t,J=5.7 Hz, 2H), 1.53(t,J=2.6 Hz, 4H);13C NMR(125 MHz, CDCl3)δ: 169.0, 158.7, 137.4, 134.8, 130.5, 130.2, 125.8, 124.5, 124.4, 124.0, 121.8, 70.9, 68.9, 38.9, 28.0, 25.2, 21.9; HR-MS(ESI)m/z: calcd for C15H19D3F3N2O2{[M+H]+}322.1822, found 322.1819。
1.3 结构表征
马来酸氟伏沙明-D3的1H NMR谱图如图1所示。谱图积分为18个氢(胺基上的两个活泼氢未出峰),与天然丰度的马来酸氟伏沙明相比,甲氧基上3个氢原子被氘取代,在高场区域未出现甲基峰,与马来酸氟伏沙明-D3的结构吻合。
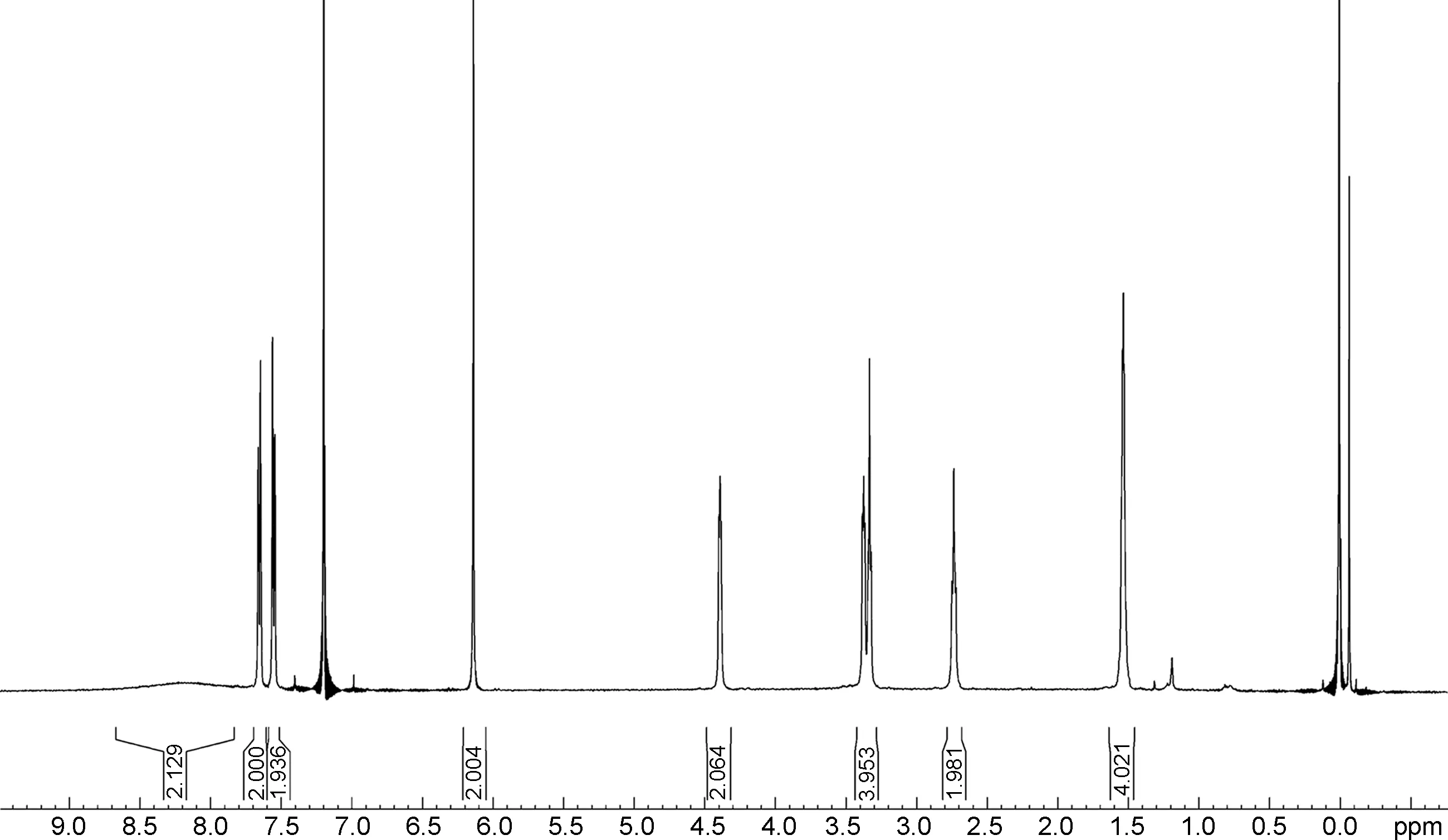
δ
马来酸氟伏沙明-D3的高分辨质谱(图2)中最高峰m/z322.1819为氟伏沙明-D3的加氢峰,即[M+H]+,其误差为0.93 ppm,结合其氢谱数、据可以确定合成的产品为马来酸氟伏沙明-D3。

m/z
5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)是合成马来酸氟伏沙明-D3的关键中间体,由4-三氟甲基苯甲醛与格氏试剂(6)经亲核加成、氧化反应制得。本文对反应过程中所涉及的溶剂温度、投料量等因素进行了考察,以确定最优反应条件。
2.1 亲核加成反应过程的优化
(1) 反应溶剂对格氏试剂制备的影响
格氏试剂的制备通常在非质子性溶剂中进行,以乙醚、四氢呋喃为反应溶剂进行考察,发现溶剂的不同对格氏试剂的生成具有直接影响。乙醚引发时间较长,如果体系无水环境控制不严格,会导致引发失败。而四氢呋喃引发速度快,加入引发剂后,保持微沸状态即可引发反应。同时,四氢呋喃的安全性更高,在工业化应用中比乙醚更具优势,因此,将四氢呋喃作为该步反应的最佳溶剂。
(2) 反应温度对亲核加成反应的影响
在格氏试剂制备完成后,考察了反应温度对亲核加成反应的影响,结果见表1。由表1可知,当反应在回流状态下进行时,收率仅有10%,说明温度过高会导致副反应增加。随着温度的降低,收率相应提高,在0 ℃下反应收率可达87%,进一步降低温度至-10 ℃,反应12 h收率出现一定下降,说明温度过低会导致反应活性的下降。最终,确定0 ℃为反应的最佳温度。
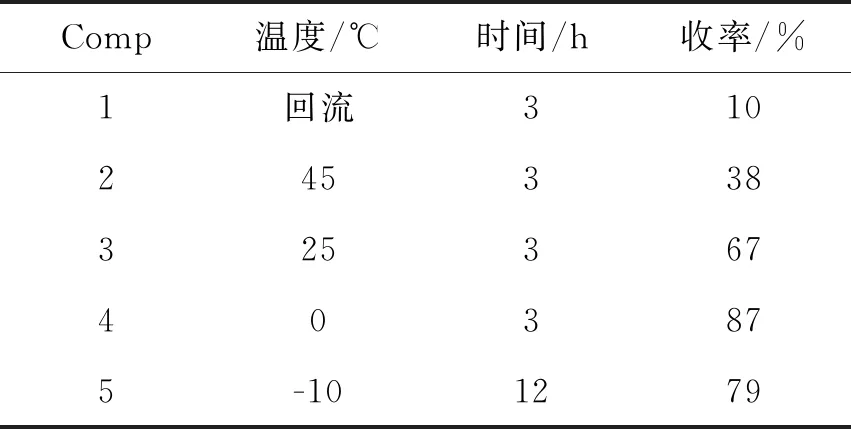
表1 温度对反应的影响
2.2 氧化过程的优化
Dess-Martin氧化反应具有反应速率快、条件温和、氧化剂用量少等特点,是现代有机合成中常用的氧化反应[17-18]。本研究首次将Dess-Martin氧化反应用于马来酸氟伏沙明-D3关键中间体9的合成中,重点从氧化剂的用量、反应溶剂等工艺参数进行了条件优化。
(1) 氧化剂用量对反应的影响
在Dess-Martin氧化反应中,氧化剂用量对氧化性能有着至关重要的作用,如表2所示,以底物5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8)为基准,当使用Dess-Martin试剂为1.0当量时,反应活性较差,反应延长至12 h,收率也仅为76%。随着氧化剂用量的增加,反应活性有明显提高,当使用1.2当量氧化剂时,收率达到了最佳值。进一步增加用量,收率再无提高。因而,最终确定Dess-Martin试剂的最佳投料量为1.2当量。

表2 氧化剂用量对反应的影响
(2) 反应溶剂对氧化反应的影响
在确定了Dess-Martin试剂的用量后,研究了溶剂对氧化反应的影响(表3)。从表3可以看出,该反应在非极性溶剂中能够得到非常好的收率,其中卤代类溶剂二氯甲烷效果最好,其次是四氢呋喃和甲苯;在极性溶剂乙酸乙酯和乙腈中反应也可以发生,但反应转化率较差。最终将二氯甲烷作为该反应的优选溶剂。
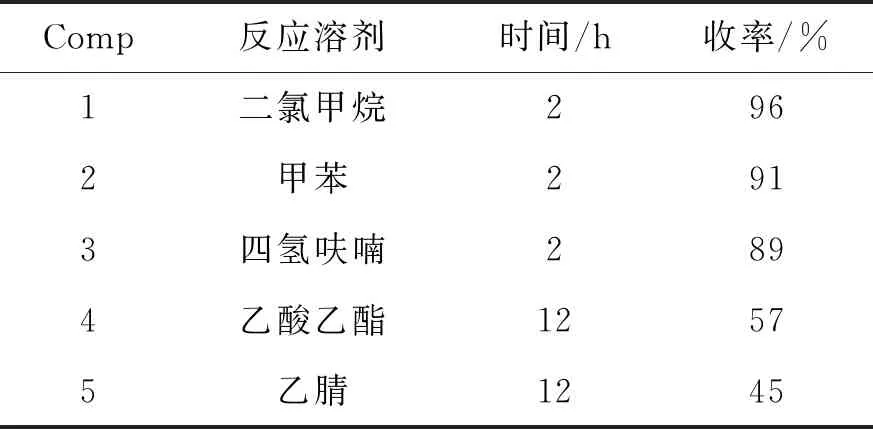
表3 溶剂对氧化反应的影响
以氘代碘甲烷为氘源,以4-苄氧基-1-丁醇(2)为起始原料,经甲基化、氢化脱苄、溴化、格氏反应、戴斯-马丁氧化、缩合、取代、成盐等反应,以51.7%总收率制备得到稳定的同位素标记的马来酸氟伏沙明-D3。其中,5-(甲氧基-D3)-1-(4-三氟甲基苯基)-1-戊酮(9)的制备是合成马来酸氟伏沙明-D3的关键步骤,本发明首次使用4-三氟甲基苯甲醛为原料与格氏试剂反应简洁高效制备出5-(甲氧基-D3)-1-(4-(三氟甲基)苯基)-1-戊醇(8),经Dess-Martin氧化得到中间体9。相对于传统方法,避免了反应周期长、重复性差及使用高毒性氰类化合物等问题。马来酸氟伏沙明-D3作为内标药物使用,可应用于临床药代动力学方面研究,从而更准确和方便地了解马来酸氟伏沙明在人体内的代谢过程和作用机制。
猜你喜欢
分子催化(2022年1期)2022-11-02
佛山陶瓷(2018年6期)2018-09-14
中国医药导报(2017年2期)2017-03-18
中国医药科学(2016年9期)2016-07-25
江苏农业科学(2014年10期)2014-11-22
职业·下旬(2014年9期)2014-10-16
中学理科·综合版(2008年11期)2008-01-14
