
含能化合物的生物合成反应
2022-10-24 11:42姜雨佳
生物加工过程 2022年5期
马 铮,姜雨佳,于 洋,李 春
(北京理工大学 化学与化工学院 生物化工研究所 医药分子科学与制剂工程工信部重点实验室,北京 100081)

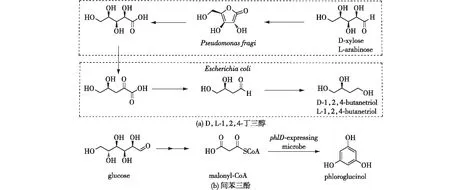
图1 D/L-1,2,4-丁三醇[7]及间苯三酚的生物合成[9]Fig.1 Biosynthesis of D/L-1,2,4-butanetriol[7] and phloroglucinol[9]
含能化合物的合成反应包括骨架的合成和修饰两个部分,其中核心反应为氮杂环和含能基团的合成。传统化学方法合成含能化合物普遍使用酸、碱、贵金属及重金属催化剂[3],导致环境污染严重,更重要的是反应过程比较剧烈、不易控制、安全性差,特别是化学催化合成的选择性差,会产生大量的副产品。由于环境问题日益受到重视,近年来绿色化学合成引起了广泛关注,研究者发展了五氧化二氮(N2O5)硝化[4-5]等绿色、高效的含能化合物化学合成工艺,比传统合成方法更加环保。
除合成化学之外,合成生物学和酶工程的发展为化学品的绿色制造提供了新思路。生物合成反应是指通过代谢酶和转运蛋白在微生物中运行的反应,或者是在体外由单个或多个酶催化的反应[6]。生物催化剂在水相、温和条件下反应,多在常温常压下进行,通常需要更少的单元操作,易于控制,而且可通过各种形式的重复发酵过程循环酶和微生物细胞,催化反应具有高度的区域选择性和立体选择性,副产物少、环境污染低,符合化学工艺绿色、安全的发展趋势。目前,在微生物细胞的代谢工程中,系统地设计和构建生物合成途径已经成为一项常规工作,以初步构建微生物菌株生产所需的产品。
1 代谢工程改造合成含能化合物前体
利用已知酶合理构建生物途径,能够合成含能化合物的前驱体。1,2,4-丁三醇是1,2,4-丁三醇三硝酸盐的前驱体。Niu等[7]通过两种起始物质D-木糖和L-阿拉伯糖,开发了一种微生物选择性合成D和L型1,2,4-丁三醇的方法(图1(a))。Feng等[8]为开发一种分离1,2,4-丁三醇的新方法,构建了水溶性差的1,2,4-丁三醇酯的生物合成途径,产物1,2,4-丁三醇可通过离心耦联水解反应回收,与传统的蒸馏、蒸发等方法相比,该工艺流程简单、能耗低,可降低工艺成本。
间苯三酚是一种用于合成1,3,5-三氨基-2,4,6-三硝基苯的前驱体,Achkar等[9]构建了以E.coliJWF1(DE3) pJA3.131A从葡萄糖生产间苯三酚的生物合成途径(图1(b))。间苯三酚对细菌有毒性,会导致重组大肠杆菌的细胞生长受到显著抑制,即使在补料发酵条件下得到的产物产量也较低,无法应用于工业生产。Zhang等[10]在大肠杆菌中过表达GroESL以期提高间苯三酚耐受性和产量,利用活细胞计数和间苯三酚产量评价菌株耐受性,结果发现,与对照菌株相比,GroESL过表达菌株的细胞活力增加3.19倍,产物间苯三酚的合成增加39.5%,达到5.3 g/L。Liu等[11]通过在E.coliBL21 (DE3)中过表达磷酸烯醇丙酮酸羧激酶或三磷酸异构酶来增加丙酮酸浓度后发现,与对照菌株相比,间苯三酚的产量提高了92%。
2 生物催化含能键的合成
虽然生物法合成含能化合物具有巨大的开发潜力,然而与合成代谢网络的理论设计取得的巨大进展相比,这种合成途径的实际实现仍然滞后。因为该领域已知酶的种类少、底物谱窄,这都受限于酶的发现和工程化改造的效率,所以已有的酶无法满足构建含能化合物合成代谢的需求[12]。目前,可以生物合成的含能化合物前驱体主要是多元醇或者酚,同时含能化合物中常出现的硝基、叠氮、偶氮等基团本身在自然界少见,但是随着天然产物的发掘,在自然界中发现了含有这些基团的分子,进而发现了催化形成这些基团反应的酶[13]。在以下部分我们将对硝基、肼、氧化偶氮和氮杂环的合成及相关酶的改造进行分类阐述。
2.1 硝化反应
生物合成的前驱体在进行硝化反应之后才能具备含能化合物的能量特性,生物硝化过程是含能化合物生物合成的首要反应。Winkler等[14]综述了硝基基团生物合成的3种机制:第一种是由一氧化氮合酶参与的催化反应;第二种是通过亲电子的硝化反应直接引入硝基;第三种是由N-加氧酶催化硝化氨基得到,这类反应通常由双核铁中心或者Rieske铁硫簇催化[15],已知的酶包括AurF、Cm1I、PmD、PvbF和SznF,其中对AurF的研究较为详细。AurF是金链菌素(aureothin)生物合成途径中的酶,对其底物谱的研究发现,AurF可以将对位是羧酸的芳香胺氧化为硝基[16]。Winkler等[17]提出,从氨基到硝基的六电子氧化分为3个两电子氧化的过程实现,通过同样的过氧-双铁中间体氧化底物,产生对应的羟胺基、亚硝基中间产物,并提出了AurF催化对氨基苯甲酸(PABA)为对硝基苯甲酸(PNBA)的反应机制(图2(a))[18]。Li等[19]提出氨基到硝基的转化是四电子氧化过程,即羟胺基到硝基的转化只需要1 mol的O2。AurF在依赖O2的氧化过程中需要NADH作为电子和氢供体以实现O2激活。与其他氧化酶类似[20],可以通过以H2O2为底物的分流途径(peroxo-shunt),以H2O2为氧供体得到过氧-双铁中间体,直接氧化氨基,从而降低反应成本[21]。
除了将氨基氧化为硝基外,对C—H的活化和直接硝化可以简单化合物为底物,省略转氨反应的中间步骤。P450单加氧酶是一类可以实现C—H键活化的血红素酶,它也被发现可以实现有机底物的硝化反应。Barry等[22]发现了一种细胞色素P450酶TxtE可以在植物毒素thaxtomin A的生物合成中直接选择性催化L -色氨酸的C4位的硝化反应。该硝化反应以一氧化氮(NO)和O2为共同底物,在体外反应中以菠菜铁氧还蛋白(Fd)和铁氧还蛋白还原酶(Fr)作为替代电子供体(图2(b))。Louka等[23]通过停流动力学方法对TxtE的反应过程进行研究后发现,O2先与NO结合,生成能够有效硝化L-色氨酸的活性铁(Ⅲ)-过氧亚硝酸盐,随后产生的NO2自由基直接硝化底物(图2(c))。同时,他们还对活性位点簇的模型进行了密度泛函理论(DFT)计算后发现,该反应过程涉及铁(Ⅲ)-过氧亚硝酸盐,其通过小的活化自由能同质分裂形成铁(IV)-氧代血红素和游离NO2自由基,NO2自由基激活底物上的芳环,而铁(IV)-氧代血红素吸收本位氢以形成产物。
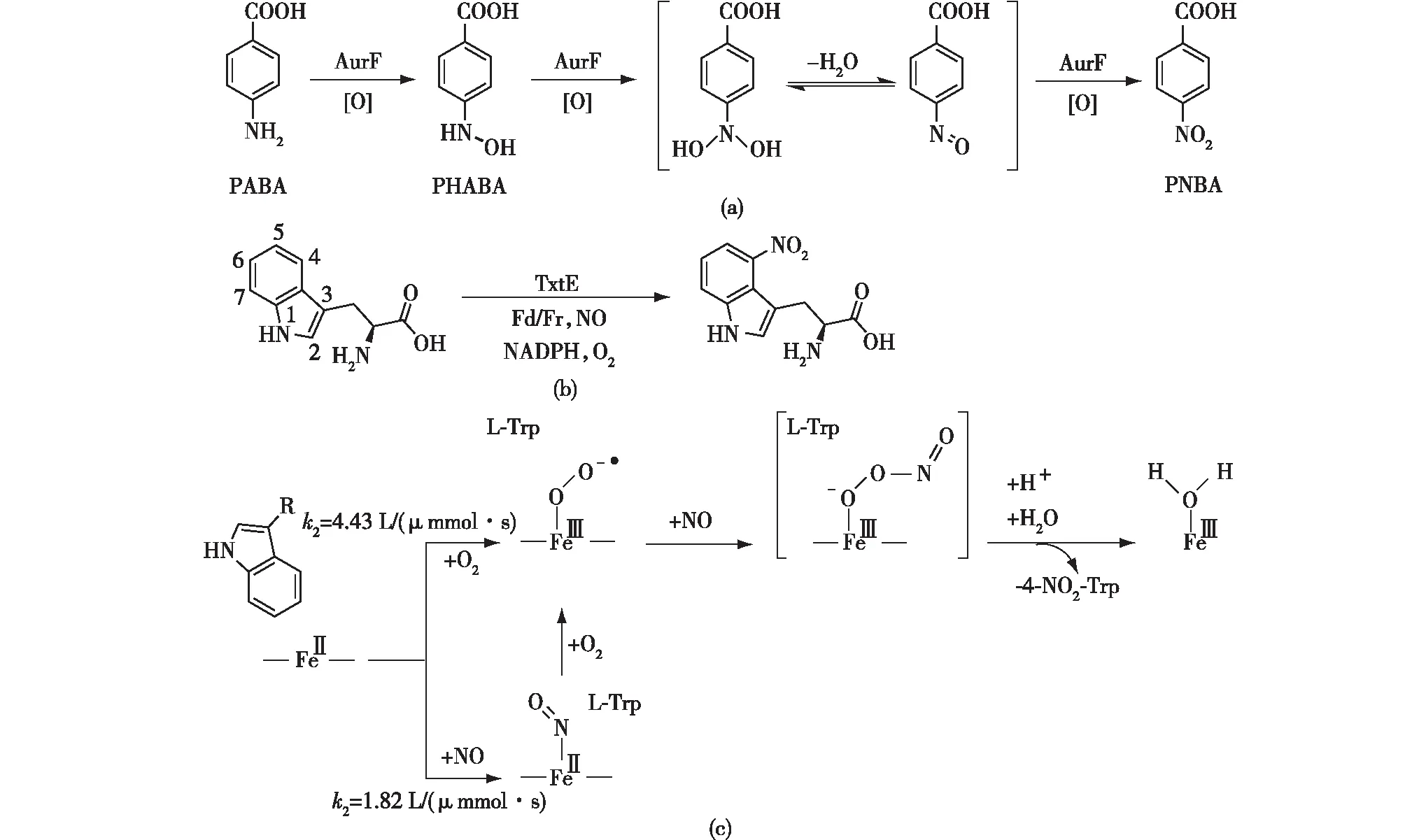
k2表示二级反应速率常数图2 AurF催化PABA的硝化反应机制[18] (a),TxtE催化L-色氨酸的硝化反应[22] (b)及TxtE的反应机制[23] (c) Fig.2 Nitration reaction mechanism of AurF[18](a),L-tryptophan nitration reactions catalyzed by TxtE[22](b),and reaction mechanism of TxtE (k2 refers to the 2nd order rate constants)[23] (c)
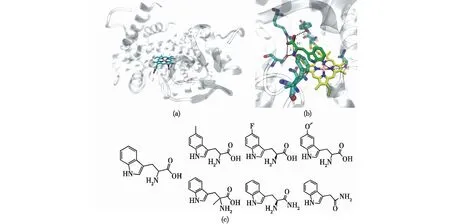
TxtE蛋白结构用灰色展示,血红素辅因子用黄色展示,L-色氨酸用绿色展示图3 TxtE的晶体结构(PDB:4TPN) (a)和TxtE的活性位点与L -色氨酸结合的晶体结构(PDB:4TPO) (b)以及TxtE可以催化的L-色氨酸结构类似的非天然底物结构(c)[24]Fig.3 Overall fold of TxtE(PDB:4TPN)(a),structure of the TxtE active site with L-tryptophan bound(PDB:4TPO)(b),and TxtE can nitrate unnatural substrates that are structurally similar to L-tryptophan (c)[24]
基于硝化酶的结构和反应机制的知识,研究者可以通过理性设计拓展硝化反应的底物谱。Dodani等[24]为了研究TxtE用于设计新的硝化生物催化剂平台的潜力,他们先获得了TxtE无底物和结合底物形式的高分辨率结构(图3(a)和(b)),并通过底物结合的光谱指示剂和底物类似物库的筛选后发现,野生型TxtE可以通过对吲哚环的中度修饰但口袋部位的氨基酸不能被修饰,从而达到对L-色氨酸结构类似的非天然底物的硝化(图3(c))。Dodani等[25]又进一步通过分子动力学模拟和Markov模型分析并结合突变设计实验发现了TxtE进行硝化反应的区域选择性开关,即通过F/G loop的单一突变可以完全将TxtE的区域选择性从L -色氨酸的C4位转移到C5位,从而仅催化产生5-硝基-L-色氨酸。这些研究表明,研究者可以通过通用的蛋白质工程的方法改造TxtE硝化酶,拓展其底物谱、改变位点选择性,为其他含能化合物的合成提高了新的选择。
与化学硝化反应相比,TxtE的酶促硝化以NO、O2为底物,利用NADPH提高驱动力,反应条件温和可控。目前研究中使用的NO来自偶氮二醇烯鎓盐类NO供体(NONOate)等小分子NO供体[24],而下一步的研究可能通过利用一氧化氮合酶、亚硝酸还原酶等以精氨酸、亚硝酸盐为底物合成NO,通过构建体外多酶催化体系,实现低成本、绿色的硝化反应。
自然界通过长期的选择进化,创造出具有高度特异性和催化能力的蛋白质,研究者们通过借鉴来自原生生物环境中的与生物硝化相关的酶(如AurF、TxtE等),对其进行设计改造,使其能够表现出所需的反应活性,并通过定向进化的方法提高这些活性。这也体现了定向进化和蛋白质工程工具在调节酶活性、选择性和稳定性以满足工业过程要求方面的重要性,也为含能化合物生物合成的工业级规模可操作性提供了条件。
2.2 肼键的合成
肼(N2H4)是一种含能物质,可用作航天器的液体推进剂。由于单质肼含能高、化学性质活泼,一直被认为不会出现在生命活动中,随着对氨分解微生物代谢的深入研究,在21世纪初,肼才在厌氧氨氧化菌中被发现并鉴定[28]。腙、酰肼及其衍生物都属于肼的衍生物,研究者陆续发现了含有腙、酰肼等结构的天然产物,进而追溯催化N—N键合成酶[13]。对此类天然产物生物合成途径的解析,尤其是涉及N—N成键机制的探究,有助于推进肼及其衍生物的高效生物合成。
2.2.1 肼衍生物的生物合成
2016年,日本东京大学的Makoto Nishiyama课题组通过探究氨基载体蛋白(Amcp)在微生物中的分布情况,在编码Amcp的链霉菌Streptomycessp.SoC090715LN-17中发现了具有独特腙单元的二肽天然产物s56-p1[29]。在2018年,该课题组的Matsuda等[30]继续研究在s56-p1生物合成途径中肼键形成的机制,并对其相关合成基因簇进行了分析鉴定,同时通过生物信息学分析发现构成这种机制的基因簇广泛存在于细菌中,表明细菌具备合成肼的潜力。然后,采用基因组挖掘的方法确定了其生物合成相关的基因簇,并将含有spb37~spb50的基因片段导入异源宿主S.lividansTK23中,在培养液的酸水解物中检测到肼的产生;再通过基因敲除确定了该化合物生物合成所需的基因为N-羟化酶的spb38和由编码Cupin和methionyl-tRNA合成酶(metRS)样蛋白结构域组成的spb40。在生物合成路径中,以L-赖氨酸为初始底物,经Spb38催化其中的末端氨基发生羟化,随后羟化的N在Spb40的催化下与甘氨酸的氨基发生反应生成肼基;N6—18OH-L-赖氨酸投喂实验的结果表明,肼键中的N—N键是通过假定的酯中间体重排得到;同时,体外活性验证实验表明,Spb38的活性在体外可以得到重建,而Spb40虽可溶性表达很好,但纯化后未表现出活性;在大肠杆菌的体内重构实验中,将spb38与spb40一起进行表达,可以在细胞代谢产物中检测到化合物1(图4)的生成。Spb39与D-氨基酸氧化酶同源,将重组Spb39加入生产化合物1的大肠杆菌过滤培养液中,可以检测到肼基乙酸(HAA)的生成,从而得出由Spb38、Spb39和Spb40介导的L -赖氨酸和甘氨酸合成HAA的生物途径(图4)。
与HAA的生物合成相似,Du等[31]发现KtzT在哌嗪酸(piperazic acid)的生物合成过程中催化了肼键的合成,在体外验证实验中,通过重组表达KtzT和与Spb38同源的L-鸟氨酸N-羟化酶KtzI,并进行耦合反应后检测到新化合物哌嗪酸的产生。该反应过程首先是通过KtzI催化伯胺的羟基化,然后哌嗪酸合酶KtzT使用血红素辅因子催化分子内N—N键的形成。
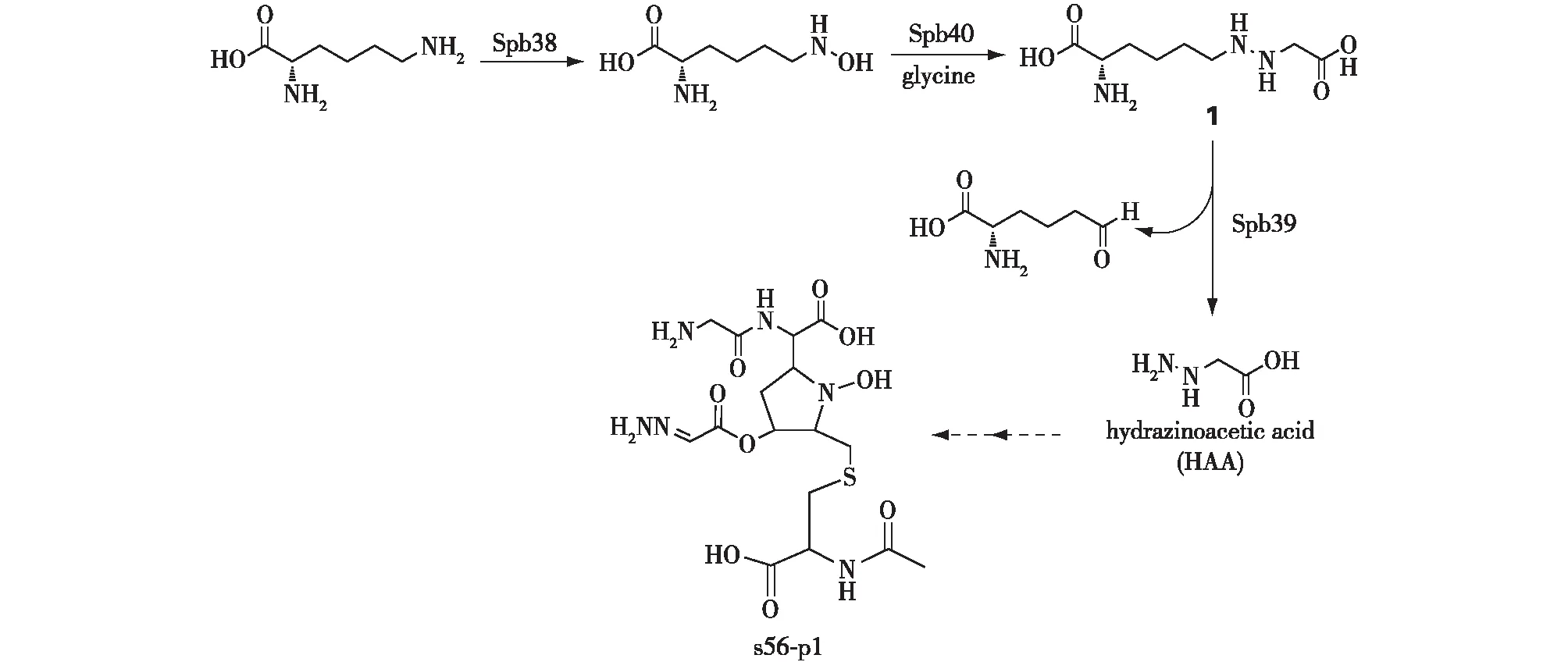
图4 肼基乙酸中N—N键的生物合成途径[30]Fig.4 Biosynthetic pathway of N—N bond formation in hydrazinoacetic acid[30]
吡唑霉素(pyrazomycin)是一种具有天然吡唑环的C-核苷类抗生素,具有与s56-p1类似的肼基结构。杜艺岭课题组的Zhao等[32]在2019年鉴定了S.candidusNRRL3601中负责吡唑霉素生物合成的基因簇;同时发现,StrR家族调控因子PyrR是控制吡唑霉素生物合成的转录激活因子。此外,该研究通过体内重建和稳定同位素喂养实验证明,PyrN是一个新的N—N键形成酶,它催化了L-谷氨酸ε-NH2和L-N6-OH-赖氨酸的α-NH2上的N形成N—N键。
Fosfazinomycin是另外一类具有肼基结构的天然产物。2015年,van der Donk课题组的Gao等[33]以膦酸酯甲基转移酶DhpI的底物耐受性作为化学选择工具,纯化得到fosfazinomycin生物合成途径中的两个中间产物,并证实了其发掘的基因簇与fosfazinomycin的形成相关。2016年,Huang等[34]对fosfazinomycin生物合成路径中相关的5种酶进行了体外活性验证并提出了其合成途径的猜想:在这项工作中发现,fosfazinomycin的N—N键似乎是在天冬氨酸(Asp)上生成的,而不是在含氮的生物合成中间体上形成的;此外,通过代谢标记研究,他们发现了这种天然产物的肼基部分中至少有1个,甚至可能2个N都来自Asp。在后续的工作中,Wang等[35]继续对该路径进行了解析,经过同位素追踪法发现,在N—N键中的1个N来自HNO2。此外,他们还发现在fosfazinomycin生物合成途径中首先生成了乙酰肼生物合成子,然后通过谷氨酰基载体将N—N键引入其生物合成途径。因此,与其他含N—N键的天然产物直接从中间体合成N—N键的合成途径不同的是,在fosfazinomycin及共相关的化合物的合成途径中,N—N键的合成途径是1个独立的路径。
2.2.2 肼的生物合成
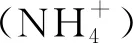
Dietl等[37]从K.stuttgartiensis中分离得到肼合酶多蛋白复合物后,成功解析了肼合酶的晶体结构,该晶体结构表明,HZS是由异源三聚体组成的狭长二聚体(α2β2γ2)蛋白,每个二聚体都含有2个c-型血红素活性位点和1个氧化还原配体的相互作用点。通过对晶体结构的分析后得出肼合酶催化肼合成的两步机制:首先,NO在γ亚基的活性位点(血红素γ-Ⅰ)经历3个三电子的还原反应,得到羟胺(NH2OH),然后,NH2OH通过蛋白内的通道系统转移到α亚基,该通道系统由β亚基内延伸的短氨基酸分支调节;最后,HZS复合物的2个血红素活性位点(血红素α-Ⅰ)促进氨(NH3)和NH2OH的缩合生成终产物N2H4。值得注意的是,该方案与工业上使用Raschig法,即氨被氧化为氯胺(NH2Cl)后,再与另1分子氨通过反歧化过程合成肼是类似的。
2.3 氧化偶氮键的合成
氧化偶氮键是高含能化合物中一种非常重要的合成组件,特别是在高能密度材料(HEDM)的合成中发挥着重要的作用,它可以通过提高材料的物理密度和能量强度、降低材料的灵敏度以及提高材料的能量水平来提高存储安全性。
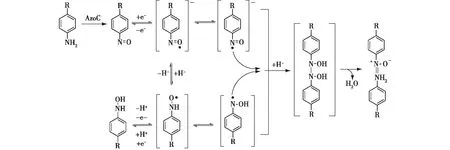
图5 氧化偶氮键的生物合成机制[41]Fig.5 Biosynthetic molecular mechanism of azoxy bond formation[41]
Valanimycin是一种含有氧化偶氮键的天然产物,于1986年从土壤微生物链霉菌S.viridifaciensMG456hF10中分离得到。Garg等[38]通过喂养实验确定了L-缬氨酸、L-丝氨酸、异丁胺和异丁羟胺是valanimycin生物合成过程中的中间体;在随后的基因簇鉴定工作中陆续发现了相应的酶:磷酸吡哆醛(PLP)依赖性的L-缬氨酸脱羧酶VlmD和黄素依赖性的异丁胺N-羟化酶VlmH,为VlmH提供NADPH的黄素还原酶VlmR,同时,valanimycin的生物合成基因簇中还特别存在1个编码丝氨酸- tRNA合成酶的基因(vlmL)。Garg等[39]根据同源序列比对后发现,VlmA与赖氨酰磷脂酰甘油合成酶具有弱序列同源性,后者是一种利用L-赖氨酰tRNA催化L-赖氨酸与磷脂酰甘油的羟基发生酰化作用的酶。
Guo等[40]于2015年从S.chattanoogensisL10中分离得到一类含氧化偶氮键的天然产物,命名为azoxymycins(氧化偶氮霉素)。随后,Guo等[41]继续解析了azoxymycins中氧化偶氮键的生物合成机制后发现,关键酶AzoC是氧化偶氮键生物合成的关键元件。他们以对氨基芳香化合物为模型,重构了氧化偶氮键生物合成的过程后发现,azoxymycins的氧化偶氮键生物合成过程是一种酶法和非酶法的级联反应:首先非血红素二铁氮氧合酶AzoC催化两步双电子氧化反应将氨基化合物转化为羟胺化合物,并继续氧化为亚硝基化合物;随后反应体系中的辅酶氧化还原对(NAD(P)+/NAD(P)H等)诱导亚硝基/羟胺化合物的快速相互转化,通过非酶促的1+1单电子转化反应生成氮原子自由基中间体,并快速二聚为氧化偶氮键(图5)。该研究团队解析了氧化偶氮键的生物分子合成机制,该研究结果为氧化偶氮化合物的生物合成提供了可能,从而为高能量密度材料氧化偶氮化合物的规模化工业生物合成提供技术基础。
目前,对含肼键和氧化偶氮键化合物的生物合成研究还集中在天然产物合成的途径解析、相关基因和酶的挖掘阶段,缺乏对酶机制的解析和后续的改造。更多肼键和氧化偶氮键合成酶的发现和机制解析将为后续的理性改造和含能化合物合成提供基础。
2.4 氮杂环化合物的合成
与传统的含能材料相比,氮杂环化合物的环结构中含有更多高能的N—N键、C—N键和更大的环张力,具有较高的生成热、更高的能量以及干净的燃烧气体(N2)的优势。
2.4.1 人工金属酶催化C—N成键
扩大蛋白质催化剂的范围,使其超越天然酶所包含的反应空间,是生物催化的重要目标和挑战。Arnold团队的Coelho等[42]和Hartwig团队的Key等[43]通过定向进化、非天然金属配位化合物引入的方法构建了人工金属酶,实现了卡宾转移反应[42-43]。在此基础上,Singh等[44]利用细胞色素P450酶通过分子内氮宾C—H插入反应,成功促进了羰酰叠氮物的活化和环化,生成恶唑烷酮这一含氮杂环化合物,这项研究揭示了细胞色素P450介导的C—H键氨化反应的机制:在催化循环中,羰酰叠氮化底物与二价铁血红素结合,形成叠氮-铁(Ⅱ)复合物,然后释放N2产生假定的亚胺-铁(Ⅳ)中间体;接着,通过逐步的氢原子捕获/自由基反弹机制进行C—H键的活化。该研究结果表明,原子经济且易获得的氮宾供体可以成功应用于催化分子内C—H键氨化的反应,也为进一步优化此类催化剂奠定了基础。
Farwell等[45]通过对细胞色素P450的活性位点进行定向进化,得到了具有对映体选择性产生氮杂环丙烷功能的新酶,与亲本酶相比,酶活性位点的突变导致叠氮化物利用率显著提高,且选择性高达99%。在用该突变酶催化对甲苯磺酰叠氮和对甲基苯乙烯生成氮杂环丙烷的反应后发现,叠氮环丙烷的形成是通过还原的二价铁血红素与对甲苯磺酰叠氮反应产生铁-nitrenoid中间体,然后与烯烃反应产生氮杂环丙烷。该研究结果表明,理性设计细胞色素P450活性位点可以实现具有挑战性的非自然反应。
Singh等[46]研究发现,细胞色素P450酶可以作为有效的催化剂,在多种芳基磺酰叠氮化合物中介导分子内苄基C—H键的胺化反应。在后续研究中,Bordeaux等[47]证明了许多含血红素的蛋白,特别是肌红蛋白和辣根过氧化物酶,是芳基磺酰叠氮C—H胺化反应的有效催化剂,他们通过对肌红蛋白突变体活性位点进行分析发现,通过蛋白质工程可以解决天然肌红蛋白骨架所提供有限的对映体选择性问题,从而产生具有改善立体选择性和对映体选择性的基于肌红蛋白的催化剂。该研究表明肌红蛋白为C—H键胺化催化剂的设计和开发提供了一个很有前途的骨架。
目前,催化控制不同的区域选择性仍然是小分子催化剂的一个挑战,而酶催化剂可以很容易通过诱变和筛选来获得所需的选择性。Arnold团队的Hyster等[48]设计了2种在C—H键胺化中具有不同区域选择性的P450BM3突变体,经研究发现,F87位的突变对于选择性的控制是重要的,F87V突变体倾向于2,5-双取代的芳基磺酰叠氮的β-胺化,而F87A突变体更倾向于α-胺化;同时发现,该酶是通过定位1个靠近铁-nitrenoid的C—H键从而实现其对于区域选择性的控制。
2.4.2 微生物从头合成氮杂环化合物
人工金属酶虽然可以实现氮杂环的一步合成,但是往往需要复杂前体。利用合成生物学方法,可以简单的底物,通过多步反应合成氮杂环化合物。吴边团队的Feng等[49]通过逆合成分析的方法设计出一条直接从基础生物原料葡萄糖到芳香类氮杂环化合物的新型人工合成途径,经过系统性的工艺优化,成功以顺次一锅发酵的方式,首次实现了高效的氮杂环微生物从头合成方法。为了突破传统的单细胞工程带来的技术障碍,他们使用3个相同物种的细胞组成了一个微生物系统,其中,氮杂环生产菌以苏氨酸为底物,通过2-氨基乙酰乙酸、1-氨基丙酮和2,5-二甲基二氢吡嗪等中间体合成2,5-二甲基吡嗪(DMP,图6),DMP再经过侧链功能化的生产菌进一步合成重要的医药中间体。通过在不同阶段依次接种功能细胞,对生物转化进行时空调整,进而模块化所需要的生物功能,实现了氮杂环分子的合成和功能化。
江会锋团队的Peng等[50]在细胞中引入甲醛酶,通过密码子优化和定向进化等方法,将乙醛转化为乙偶姻的效率提高40倍,以1.5 mol/L的乙醛为底物积累了56.7 g/L的乙偶姻;在连续补料条件下,乙偶姻的质量浓度可达222 g/L,乙醛转化率为85%;在得到乙偶姻后,通过乙偶姻与铵盐的体外自发反应可以得到94 g/L川芎嗪,乙醛转化率为48%。这一工作也展示了生物-化学联合转化实现氮杂环化合物分子合成的潜力。
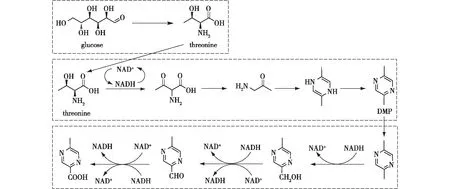
图6 微生物协同全合成功能性氮杂环[49]Fig.6 Cooperative microbes for the total biosynthesis of functionalized N-heterocycles[49]
3 设计指导含能化合物的生物合成
含能化合物生物合成的研究主要集中于前驱体合成和生物硝化两方面,虽然取得了一定的进展,但目前还未达到能够在生物体内从头合成已知含能化合物的水平。而对于新的性能良好含能化合物的直接生物合成还处于理论路线设计层面。与研发含能化合物相比,生物合成技术的挑战来源于多方面,如:含能化合物中含有的硝基、叠氮和偶氮等基团很难通过生物催化引入;需要考虑到在生物体内进行合成时,生物体的耐受能力以及后续的终产物分离纯化过程等。
随着高性能计算技术的迅猛发展,基于生物平台的绿色合成制造技术朝着更高效、更智能、更具有创造性的方向进发。定向进化和新兴的计算设计都是含能化合物生物合成的重要研究方法。
酶的生物转化具有非常广泛的底物范围和高的反应性和选择性,这种生物催化系统使我们能够轻易地获得在自然界中很少见到的多功能分子结构,并扩展生物系统可及的化学空间。高应变碳环是一类高能量化合物,双环丁烷折叠的结构形式是张力最大的四元体系之一,应变为276.3 kJ/mol[51]。Arnold研究团队的Chen等[52]通过定向进化改造P450酶,成功构建了用于生产高应变双环丁烷和环丁烯的生物催化平台。该蛋白通过激活铁-卡宾体使卡宾加到炔烃上,在双环丁烷生成中稳定反应性的环丙烯中间体以及卡宾转移过程的精确立体控制,从而实现了所需的反应转化。他们首先发现丝氨酸配位的P411血红素蛋白具有卡宾转移炔烃的潜力,然后通过分析设计得到突变体P411-E10(P4 A78V A82L F263L),发现E10能够催化产生双环丁烷,接着选择P411-E10突变体作为起始模板用于定向进化,成功构建出一种更高效且底物谱更广泛的双环丁烷酶和环丁烯酶。
创新性引入生物催化过程为绿色化学的应用提供了广阔的前景。然而由于有限的催化性能,从自然界生物多样性中获得的酶往往需要通过长时间、反复的实验室进化来改进才能获得所需的功能。Das等[53]和Richter 等[54]从蛋白质折叠算法出发,开发了Rosetta程序包,可以实现多种蛋白质的设计功能[53-54],他们通过Rosetta设计了新酶,催化Kemp消除[55]和反醇醛缩合[56]等已知天然酶不能催化的反应。基于Rosetta及其他蛋白质设计方法,研究者可以优化已知人工金属酶的结构,提高催化活性[57];也可以设计新酶,实现手性β-氨基酸[58]和D-氨基酸等实验室合成和工程应用[59]。
4 总结与展望
含能化合物由于自身结构性能特点,在制备过程中存在高危易爆的风险。除了追求性能更高、更安全的含能材料之外,含能化合物制造工艺的安全环保也是重要的目标。生物合成含能化合物作为代表性的绿色化学合成方法,秉承可再生可持续发展的理念,具有从可再生资源中提取并使用可持续方法制造的特点,是一种有前景、有潜力、可发展推广的新型技术。酶作为催化剂,能够加速化学转化的数量级,同时表现出可控制的选择性,但存在于狭窄的底物范围和有限的反应类型等缺点。而近年来发展的人工金属酶(ArM)技术在克服这些挑战方面显示出了潜力,随着结构生物学的发展,ArM的研究进展迅速。通过理性设计改造,ArM可以催化天然酶无法进行的非自然反应,而且具有高反应选择性、宽泛的底物谱及生物相容性等特点。通过将ArM催化引入含能化合物生物合成的代谢途径中,将会使该生物合成过程更高效、更易于设计且具有更高的可控性。
随着微生物组学、代谢工程和合成生物学领域的快速发展,我们已经有能力构建具有特定功能的微生物,并能合成大量具有医学和工业重要性的化合物。通过代谢工程可以使药物、化学品、燃料和材料的生物生产成为可能并以此发展起来,通过合成生物学可以建立生物部件、模块和系统来理解和操纵生物工厂。为了实现含能化合物的生物全合成,生物合成机制解析、代谢途径构建、细胞产物释放、产物分离纯化等方面均要给予考虑。为了实现含能化合物生物全合成的工业级生产,除了生物催化在上游代谢途径的优化之外,该工艺的下游提取过程也应当符合“绿色化学”要求。利用微生物细胞代替化学工厂合成含能化合物具有广阔的应用前景,在今后的发展过程中,基于生物平台的绿色合成方法不仅会成为能源材料工业,而且会成为整个化学工业高度追求的目标。
猜你喜欢
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
化工管理(2020年1期)2020-01-16
世界农药(2019年4期)2019-12-30
肉类研究(2019年8期)2019-09-10
天然产物研究与开发(2018年10期)2018-11-06
科技资讯(2018年16期)2018-10-26
科技资讯(2017年12期)2017-06-09
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
