
高效液相色谱法测定维生素D3的三个标准方法比较
2022-10-25 07:57秦天琦吴愫青广东省珠海市食品药品检验所广东珠海519090
首都食品与医药 2022年20期
秦天琦,吴愫青(广东省珠海市食品药品检验所,广东 珠海 519090)
碳酸钙D3片为复方制剂,含有碳酸钙和维生素D3两种主要的有效成分。钙元素是维持人体神经、肌肉、骨骼系统、细胞膜和毛细血管通透性等正常生理功能所必需的。维生素D3是自然存在的一种脂溶性维生素,属于类固醇类激素,维生素D3的基本功能是参与体内钙、磷代谢,促进肠道钙、磷吸收,调整钙、磷吸收比例,促进骨的钙化和正常发育[1]。碳酸钙D3片临床上主要用于妊娠和哺乳期妇女、更年期妇女、老年人等的钙补充,并用于预防与治疗佝偻病、中老年骨质疏松症,另有文献报道维生素D3对老年高血压亦有协同治疗作用[2]。
因制剂中维生素D3含量较低,准确测定制剂中维生素D3的含量尤为重要,目前已有的三个标准:《国家食品药品监督管理局标准YBH05962009》《国家食品药品监督管理局国家药品标准WS1-(X-002)-2008Z-2012》《GB 5009.82-2016食品安全国家标准 食品中维生素 A,D,E的测定》,均对如何提取分离并准确测定制剂中的维生素D3作了相关的规定,本文参考三个标准的规定,分别采用萃取后真空旋蒸至干复溶法、有机溶剂提取法、皂化法三种方法提取分离碳酸钙D3片中的维生素D3,然后分别采用反相高效液相色谱法、正相高效液相色谱法、在线固相萃取-二维液相色谱方法三种方法测定维生素D3的含量。
1 仪器与试药
1.1试剂与药品 药品:碳酸钙D3片(北京振东康远制药有限公司提供;批号:20201056),对照品:维生素D3(中检院提供,批号:100061-201208,含量:99.8%,干燥:无),试剂:异丙醇、氨水、无水乙醇、环己烷、无水硫酸钠、氯化钠、乙腈(HPLC)、甲醇(HPLC)、二甲基亚砜、正己烷(HPLC)、异丙醇(HPLC)、抗坏血酸、2,6-二叔丁基对甲酚(简称BHT)、氢氧化钾。
1.2仪器与色谱柱 仪器:电子分析天平(METTLER TOLEDO XPE206DR);超高效液相色谱仪(Agilent 1260 prime);高效液相色谱仪(Agilent 1260);超声波清洗器(Elma P300H);水浴锅(Julabo CORIO CD);低速台式离心机(Anke TDL-50C);旋转蒸发仪(IKA RV10 Auto pr)。
色谱柱:反相色谱法色谱柱:Welch Ultimate LP-C18,4.6×150mm,5μm,SN:60201 201105;正相色谱法色谱柱:Welch Ultimate SiO2,4.6×250mm,5μm,SN:60210 301071;在线固相萃取-二维液相色谱方法色谱柱:SPE柱:Agilent PLRP-S,4.6×12.5mm,15-20μm(5982-1270);1D分析柱:Agilent Poroshell 120 EC-C8,4.6×100mm,4 µm(695970-906);捕获柱:Agilent Poroshell 120 EC-C18,4.6×5mm,4 µm(820750-916);2D分析柱:Agilent Zorbax Eclipse Plus PAH,2.1×100mm,3.5 µm(959793-918)。
2 实验部分
2.1提取分离维生素D3
2.1.1萃取后真空旋蒸至干复溶法 系统适应性试验:维生素D3峰与前维生素D3峰的分离度应符合规定。
前维生素D3相对校正因子测定:避光操作。取维生素D3对照品约20mg,精密称定,置200ml量瓶中,加异丙醇溶解并稀释至刻度,摇匀,作为对照品贮备溶液,精密量取5ml,置100ml量瓶中,加异丙醇至刻度,摇匀;另精密量取对照品贮备溶液5ml,置250ml回流瓶中,加异丙醇20ml,于95℃水浴中回流1小时,40℃真空旋转蒸发至干,精密加入异丙醇100ml,振摇使溶解。精密量取上述二种溶液各20μl注入液相色谱仪中,计算前维生素D3折算成维生素D3的相对校正因子(f=(A1-A2)/(A3-A4))。
式中:A1为热破坏前维生素D3的峰面积;A2为热破坏后维生素D3的峰面积;A3为热破坏后前维生素D3的峰面积;A4为热破坏前前维生素D3的峰面积。
供试品溶液:避光操作。取本品1片,置250ml具塞量筒中,加0.3%氨水溶液,振摇使崩解,置90℃-100℃水浴中,加热振摇15秒,取出,具塞,振摇45秒,再重复上述操作2次。加无水乙醇至60ml,振摇10秒钟,精密加入环己烷100ml,振摇1分钟,加水50ml,振摇2分钟,静置使分层(若乳化,可加入氯化钠适量破乳),取环己烷层,经无水硫酸钠滤过,精密量取续滤液50ml,置150ml回流瓶中,40℃真空旋转蒸发至干,精密加入异丙醇2ml,振摇使溶解。
对照品溶液:精密称取维生素D3对照品适量,加异丙醇溶解并定量稀释制成每1ml中约50IU的溶液,同法测定。
2.1.2有机溶剂提取法 系统适应性试验:维生素D3的拖尾因子不得大于2.0。取对照品溶液重复进样6次,维生素D3峰面积的相对标准偏差应不大于3.0%;另取对照品储备液Ⅱ适量,在55℃水浴中加热1小时,放冷,取该溶液40μl注入液相色谱仪,维生素D3前体峰与维生素D3峰之间的分离度应大于3.0。
供试品溶液:取本品20片,精密称定,研细,精密称取适量(约相当于维生素D3250IU),置50ml离心管中,加入75%二甲亚砜水溶液约20ml,密塞摇匀,将离心管置于45℃±5℃水浴超声15分钟,并不时振摇,放冷。精密加入用75%二甲亚砜水溶液处理过的正己烷(75%二甲亚砜水溶液和正己烷按1∶20进行混匀后,取正己烷层)15ml,振摇90分钟,3000rpm离心10分钟,取上清液作为供试品溶液。
对照品溶液:精密称取维生素D3对照品(40000IU/mg)约25mg,置100ml量瓶中,加上述75%二甲亚砜水溶液处理过的正己烷溶解并稀释至刻度,摇匀,作为对照品储备液Ⅰ。精密量取对照品储备液Ⅰ5ml,置100ml量瓶中,加上述75%二甲亚砜水溶液处理过的正己烷溶解并稀释至刻度,摇匀,作为对照品储备液Ⅱ。再精密量取对照品储备液Ⅱ5ml,置100ml量瓶中,加上述75%二甲亚砜水溶液处理过的正己烷溶解并稀释至刻度,摇匀,作为对照品溶液。
精密量取供试品溶液和对照品溶液各40ml,分别注入液相色谱仪,记录色谱图。取上述75%二甲亚砜水溶液处理过的正己烷作为空白溶液,同法进样,以消除对含量测定的干扰。按下式计算维生素D3的总量:维生素D3总量=维生素D3含量+维生素D3前体含量=维生素D3含量×1.09。
2.1.3皂化法 供试品溶液:取本品1片置于圆底烧瓶中,加入20ml的水振摇使溶解,混匀,再加入1.0g抗坏血酸和0.1gBHT,混匀,加入30ml的乙醇和10-20ml的氢氧化钾溶液(50g KOH/50g水),混匀后于80℃恒温水浴皂化30min。皂化后冷却至室温,用乙醇∶水=1∶1转移定容至100ml。取适量皂化液,先高速离心10min(>5000rpm),再用0.22μm的针式滤器过滤(尼龙滤膜,5190-5271)至进样瓶中,待测。
对照品溶液:精密称取维生素D3标准品10.0mg,用无水乙醇溶解后,转移至100ml容量瓶定容,精密量取0.5ml于100ml容量瓶中,用甲醇定容至刻度,再精密量取1ml于10ml容量瓶中,用甲醇定容至刻度,摇匀(VD3:0.05μg/ml)。
2.2维生素D3含量测定
2.2.1反相高效液相色谱法 采用Agilent超高效液相色谱仪,以十八烷基键合硅胶为填充剂(色谱柱: Welch Ultimate LPC18,4.6×150mm,5μm,SN:60201201105),以乙腈-甲醇(9∶1)为流动相,流速为1 ml/分钟,检测波长为265nm,进样量为20μl,记录色谱图。
2.2.2正相高效液相色谱法 采用Agilent高效液相色谱仪,以硅胶为填充剂(色谱柱: Welch Ultimate SiO2,4.6×250mm,5μm,SN:60210301071),流速为1.0 ml/分钟,检测波长为265nm,进样量为40μl,以含0.45%(V/V)异丙醇的正己烷溶液为流动相A,以含20%(V/V)异丙醇的正己烷溶液为流动相B,按表1进行梯度洗脱,记录色谱图。
2.2.3在线固相萃取-二维液相色谱方法 采用Agilent高效液相色谱仪,按下面步骤确定维生素D3分析方法。
第一步:流路连接,见图1。
第二步:确定维生素D3的切割窗口。根据色谱条件(表2)按顺序分别进一针空白溶液,两针对照品溶液,保证保留时间重复性后,得到维生素D3的保留时间。
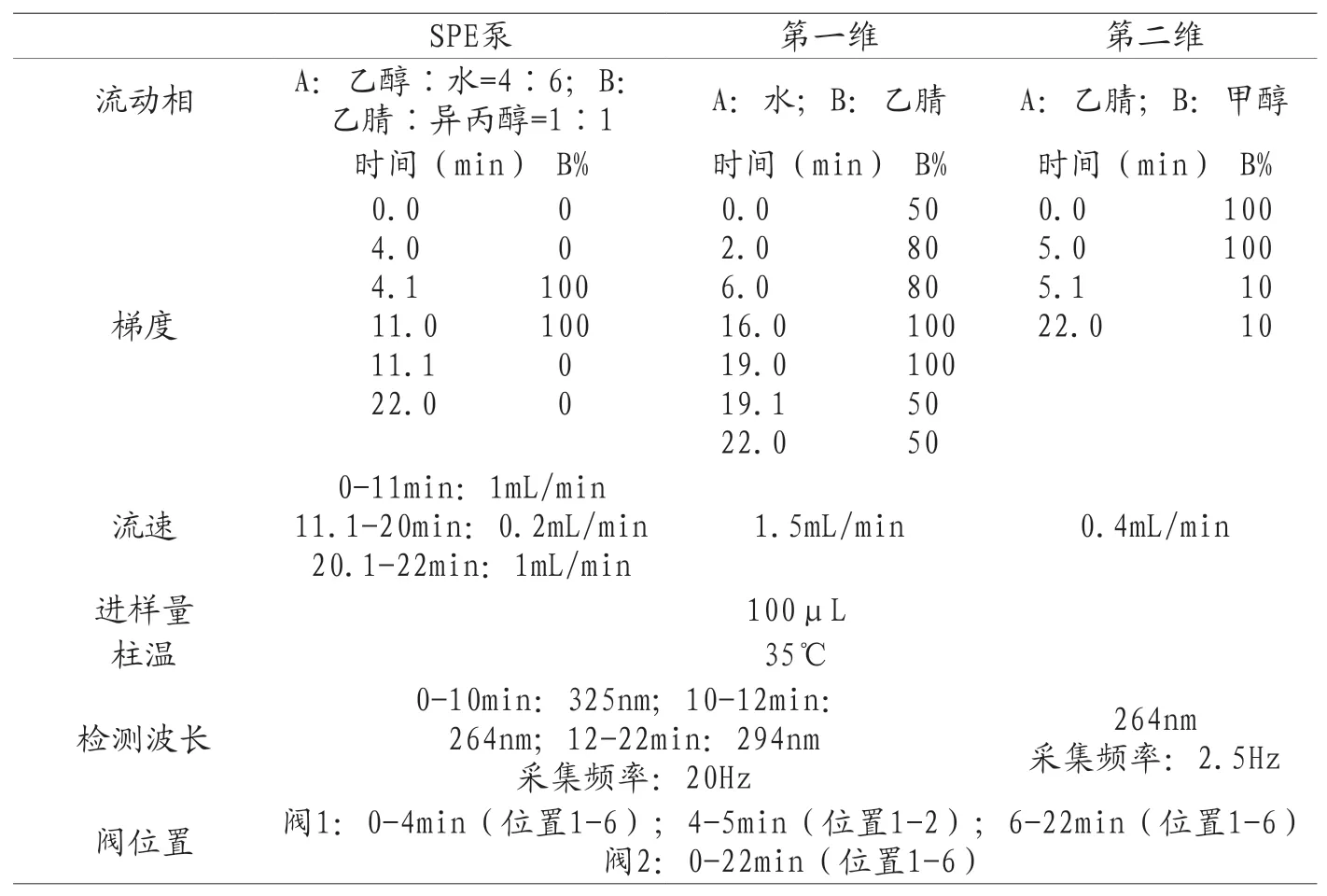
表2 维生素D3系统验证分析方法
第三步:建立完整的维生素D3分析方法。根据结果确定维生素D3捕获窗口并编辑阀2的切换时间。切换开始时间为维生素D3峰起始时间减去0.1min,切换结束时间为维生素D3峰回到基线时间加0.1min。其余条件与表2条件一样,得到完整的维生素D3分析方法(见表3所示)。用该方法再进一针对照品溶液,可以在1D检测器和2D检测器分布得到维生素D3色谱图。
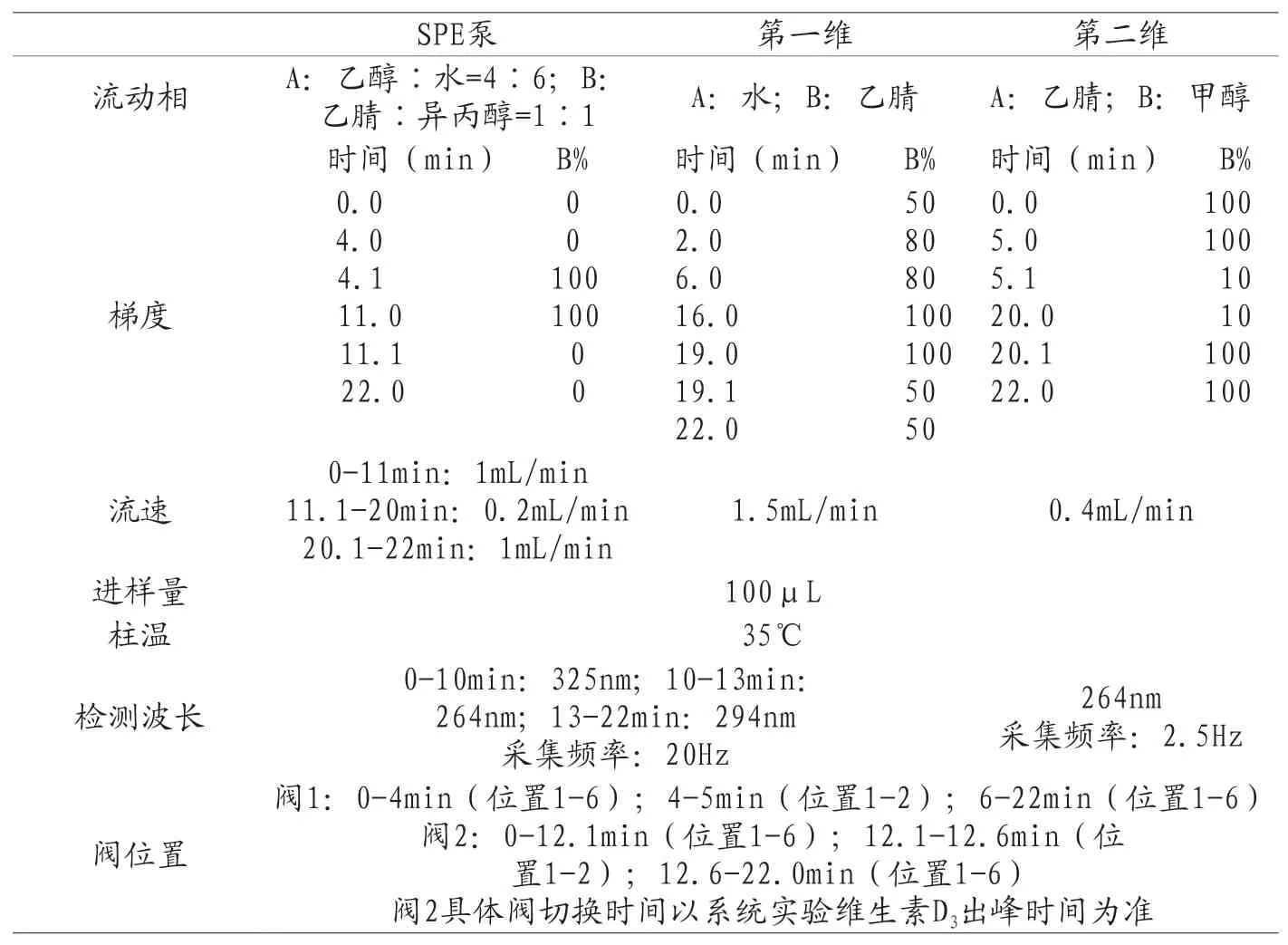
表3 维生素D3完整分析方法
3 实验结果分析
3.1萃取后真空旋蒸至干复溶法-反相高效液相色谱法
3.1.1系统适用性 按2.1.1所述方法,制备热破坏后的维生素D3对照品溶液,精密吸取溶液按照2.2.1所述色谱条件进样测定,色谱图见图2。结果表明,维生素D3峰(保留时间17.531min)与前维生素D3峰(保留时间16.472min)的分离度符合药典规定的要求(1.6)。
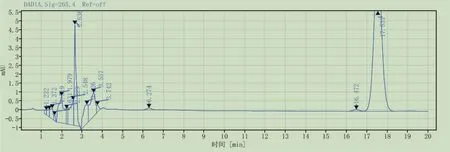
图2 系统适用性色谱图
3.1.2含量测定结果 取供试品共10份,依法测定结果,并计算相对平均偏差。10份样品的平均含量为108.6%,在药典规定的范围内(90.0%-122.5%),相对平均偏差为1.8%。
3.2有机溶剂提取法-正相高效液相色谱法
3.2.1系统适用性 按2.1.2所述方法,制备系统适用性溶液,精密吸取系统适应性溶液,按照2.2.2所述色谱条件进样测定,色谱图见图3。结果表明,维生素D3的拖尾因子不大于2.0(1.3),取对照品溶液重复进样6次,维生素D3峰面积的相对标准偏差不大于3.0%(0.4%),维生素D3前体峰(保留时间16.733min)与维生素D3峰(保留时间28.004min)之间的分离度大于3.0(17.2)。
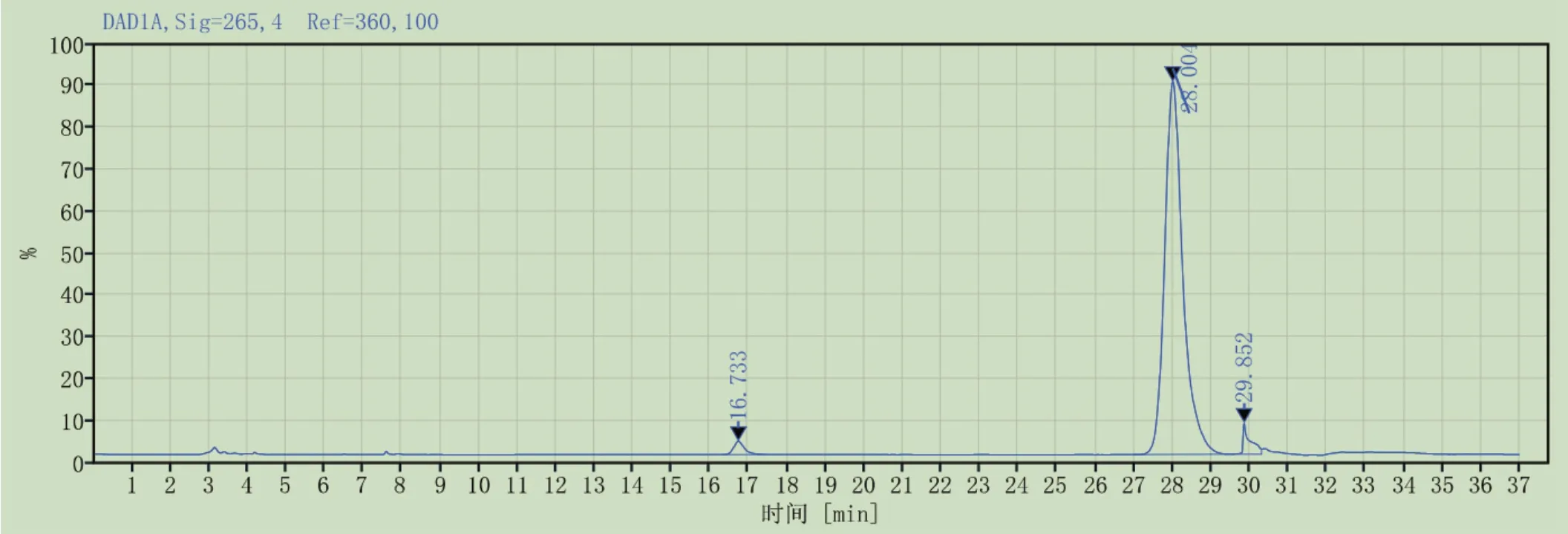
图3 系统适用性色谱图
3.2.2含量测定结果 取供试品共2份,依法测定结果,并计算相对平均偏差。2份样品的平均含量为102.3%,在药典规定的范围内(90.0%-122.5%),相对平均偏差为0.6%。
3.3皂化法-在线固相萃取-二维液相色谱方法
3.3.1系统适用性 按2.1.3所述方法,制备对照品溶液,精密吸取对照品溶液,按照2.2.3所述色谱条件进样测定,色谱图见图4。结果表明,维生素D3出峰时间为21.899min,且与相邻峰分离良好。
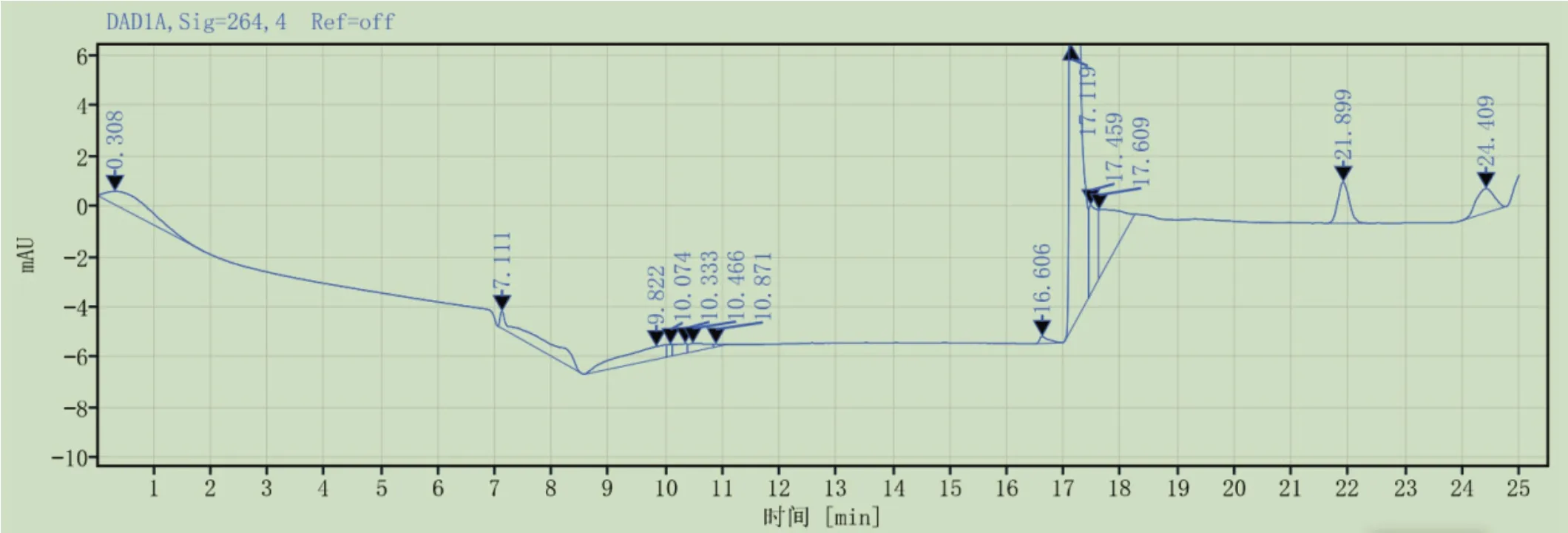
图4 系统适用性色谱图
3.3.2测量线性与范围 按2.1.3所述方法,制备不同浓度的对照品溶液(0.005μg/ml、0.0125μg/ml、0.025μg/ml、0.05μg/ml、0.125μg/ml、0.25μg/ml),以测得的响应信号(峰面积)对被分析物浓度的函数作图,用最小二乘法进行线性回归,结果如图5所示。结果表明,线性相关系数r2=0.993,线性关系良好。
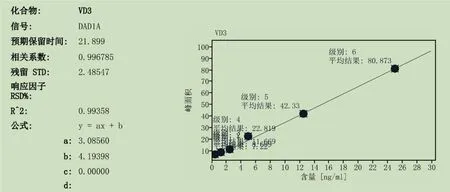
图5 维生素D3含量测定线性范围
3.3.3含量测定结果 取供试品共2份,依法测定结果,并计算相对平均偏差。2份样品的平均含量为103.5%,在药典规定的范围内(90.0%-122.5%),相对平均偏差为1.0%。
4 讨论
碳酸钙D3片中维生素D3含量很低,一直以来其分析过程操作繁琐,样品回收重现性差。碳酸钙D3片是临床大量使用的一种钙补充剂,对其准确定量意义显著。本文采用了三种方法提取分离并测定碳酸钙D3片中的维生素D3含量,三种方法测定的含量结果分别为108.6%、102.3%、103.5%,均在药典规定的范围内,三种提取方法均满足对碳酸钙D3片中维生素D3测定的前处理要求,其中真空旋蒸至干复溶法,提取过程复杂,耗时费力,有机溶剂提取法,耗费大量有机试剂,和目前提倡的环境保护理念不相适应。而本文中使用的皂化法前处理步骤最方便快捷,相对于《GB 5009.82-2016食品安全国家标准 食品中维生素 A,D,E的测定》中采用的提取方法,简化了提取步骤,避免了大量破坏环境有机试剂的使用,配合使用在线固相萃取-二维液相色谱方法测定含量,效率更高、准确度更好,此方法更适用于碳酸钙D3片中维生素D3含量的测定。
近年来,在线固相萃取-二维液相色谱技术以峰容量大、基质效应低、分析通量高等特点被广泛应用于食品检测行业中[3],但在药物检测中的应用相对较少。《中国药典》2020年版四部(通则0722)对维生素D的前处理须通过皂化、提取、分离、液相收集等步骤,这种前处理方式较为繁琐,不适合处理大量样品,而且对实验人员的操作水平也有较高要求。本文中采用的分析方法将样品溶液皂化后经简单定容便可直接进行检测,进样后样品溶液经第一个六通阀,通过在线固相萃取小柱净化,净化液进入一维色谱,一维色谱采用C8色谱柱,进行初步分离,根据维生素D3在一维色谱上的出峰时间,确定切换时间,将这一段时间内的流出液经另一个六通阀上的捕获柱富集,转入二维色谱进一步分离,实现对维生素D3的快速测定[4-5]。此方法不仅可以用于制剂中维生素D3的检测,还可以用于复合维生素片中维生素A、维生素D2、维生素D3、维生素E的分离检测。在线固相萃取-二维液相色谱技术作为一种高效的分离分析手段,与传统的定量方法相比,该柱切换系统无需繁琐的制备过程,大大提高了分析效率,该技术在婴幼儿配方食品、配方乳制品中维生素A、D、E的实际检测应用方面已经取得了较好的效果,也为复合维生素片中维生素A、D、E的含量测定提供了新思路和新参考。但是,二维液相色谱仪器的价格昂贵且维护成本较高,导致该技术在维生素A、D和E检测中的应用方面受到了局限,多用于研究性领域,另外对于复杂制剂中维生素A、D、E和其他脂溶性维生素的检测还有待于更深入的研究和开发。未来,随着二维液相色谱的不断发展完善,以及应用面的扩大和经验的积累和总结,这一技术将具有良好的应用前景。
猜你喜欢
当代水产(2022年4期)2022-06-05
ELLE世界时装之苑(2021年10期)2021-10-11
科学与财富(2021年34期)2021-05-10
中国药学药品知识仓库(2021年18期)2021-02-28
分析化学(2018年4期)2018-11-02
分析化学(2017年12期)2017-12-25
电子技术与软件工程(2016年24期)2017-02-23
食品界(2016年10期)2016-09-10
中国医药科学(2015年4期)2015-05-20
新媒体研究(2014年10期)2014-06-26
