
基于处方挖掘与分子动力学模拟筛选严重急性呼吸综合征冠状病毒2潜在抑制剂分子的研究*
2022-10-25 04:20张晓铮邰杨浩梁力中张玉龙侯淑琳
生物化学与生物物理进展 2022年10期
张晓铮 高 颖 刘 玉 邰杨浩 梁力中 张玉龙 侯淑琳 解 军**
(1)山西医科大学基础医学院,生物化学与分子生物学教研室,出生缺陷与细胞再生山西省重点实验室,太原 030001;2)山西医科大学第二临床医学院,太原030001;3)山西医科大学第一临床医学院,太原 030001;4)中国科学院广州生物医药与健康研究院,呼吸疾病国家重点实验室,广州 510530)
严重急性呼吸综合征冠状病毒2(SARS-CoV-2)引起的新型冠状病毒肺炎(COVID-19)于2019年12月31日被首次发现[1],其在全球范围内的广泛传播对全人类的生命健康造成了严重危害。截至2022年4月,全球已有超过5.14亿人确诊,625万余人死亡。COVID-19患者以发热、干咳、乏力为典型症状,重症患者可出现急性呼吸窘迫综合征、脓毒症休克、多器官功能衰竭等症状[2]。SARSCoV-2表面刺突蛋白(spike protein,S蛋白)的受体结合域(receptor binding domain,RBD)能够与人体细胞的血管紧张素转化酶2(angiotensinconverting enzyme 2,ACE2)结合,这是病毒感染人体的主要途径[3]。
目前尚无SARS-CoV-2特异性抗病毒药物,临床治疗以对症治疗和支持治疗为主[4]。现有西医药疗法约占临床试验数的40.9%,涉及西药种类52种,其中包括洛匹那韦/利托那韦、达芦那韦/考比司他、抗流感药物法匹拉韦、巴洛沙为酯和奥司他韦等。洛匹那韦和利托那韦均为HIV-1蛋白酶抑制剂,而瑞德西韦[5]的作用机制是整合进SARSCoV-2的RNA链中,阻断RNA复制,最终导致RNA链终止延迟。临床研究显示,洛匹那韦[6]等西药对COVID-19有一定抑制作用,但存在胃肠道反应、肝功能损害、脂质代谢异常等不良反应。同时瑞德西韦[7]、洛匹那韦和利托那韦[8]等可能无法进入细胞抑制病毒复制、装配,导致这些药物单独使用效果较差。抗体药物疗法约占临床试验数的19.2%,其优点在于特异性强、安全性好等[9]。中国科学院微生物研究所从康复患者体内分离出一株高效单克隆抗体——埃特司韦单抗(Etesevima),其能有效预防和治疗COVID-19。Etesevima与Bamlanivimab双抗体疗法效果更佳,但是对奥密克戎(Omicron)变异株失效[10]。美国再生元制药公司和瑞士罗氏制药公司联合开创了一种新的抗体疗法,该疗法由伊维德单抗(REGEN10987)和卡西里单抗(REGEN10933)组成[11]。该抗体主要适用于轻中度患者,但对奥密克戎变异株失去抑制作用[12-14]。值得注意的是,这两种抗体疗法都是通过非竞争性结合S蛋白的RBD结构域,阻止S蛋白RBD与人体ACE2受体结合,进而避免病毒感染宿主细胞[11]。因此,抑制SARS-CoV-2与ACE2受体的结合,进而阻止病毒与宿主细胞的结合在预防和治疗COVID-19中发挥重要作用。但目前候选抗体药物多数从康复患者血清中获取,来源非常有限;且有研究表明,SARS-CoV-2可能导致抗体依赖 性 增 强 (antibody-dependent enhancement,ADE)效应[15],无法避免其在宿主体内的核酸复制和装配。
中医药疗法约占COVID-19治疗性临床试验的39.9%,在COVID-19的预防和临床治疗过程中发挥了积极作用[16]。中国国务院新闻办在《新型冠状病毒肺炎诊疗方案(试行第八版)》中推荐使用金花清感颗粒、连花清瘟胶囊(颗粒)、清肺排毒汤等进行治疗[2]。以清肺排毒汤为例,其主要中药成分如槲皮素、柚皮素、木犀草素通过作用于多个靶点,涉及多种信号通路,参与调控机体代谢、免疫调节、炎症反应等生理过程,呈现出抗炎、抗病毒的作用[17]。然而,这些中药处方中对COVID-19治疗起到关键作用的活性成分以及这些活性成分抗SARS-CoV-2的分子机制尚不明确,这在很大程度上阻碍了中医药在COVID-19治疗中的应用。因此,筛选COVID-19治疗处方中的有效活性成分并阐明其抑制SARS-CoV-2感染的内在分子机制对开发治疗COVID-19效果显著的药物意义重大。
本文首先通过检索COVID-19治疗中药处方,挖掘潜在SARS-CoV-2抑制活性成分,构建了“新冠肺炎中药候选活性成分数据库”。基于ACE2与SARS-CoV-2表面S蛋白的结合位点已成为COVID-19的潜在药物作用靶点[18]。本文以具有人源ACE2抑制作用的活性小分子化合物为训练集构建药效团模型,并以优选药效团模型对“新冠肺炎中药候选活性成分数据库”进行虚拟筛选,得到能与ACE2结合的潜在抑制SARS-CoV-2活性成分。随后,本文将上述潜在抑制活性成分作为配体,与人源ACE2受体蛋白进行分子对接,预测了10个可与ACE2/SARS-CoV-2识别位点结合的中药成分。最后,通过分子动力学模拟探究了具有潜在SARSCoV-2抑制作用的活性小分子与ACE2蛋白的互作机制,进一步从原子水平揭示了其抑制SARSCoV-2 S蛋白RBD与ACE2结合的内在机制。本项研究将为SARS-CoV-2特异性抗病毒药物的研发提供必要的理论指导。
1 材料与方法
1.1 中药活性成分获取
1.1.1 中药处方挖掘
研究基于已报道的用于临床治疗的处方,同时参考中国各地方针对COVID-19治疗的不同处方以及已公布的《新型冠状病毒肺炎诊疗方案(试行第八版)》中的中药复方,全面搜集抗新冠肺炎的有效方剂,重复者只取其一。
1.1.2 COVID-19中药候选活性成分数据库的建立
参考《中华本草》和《中国药典》对处方中的中药进行名称规范及频数统计,取频次≥5的中药材作为研究对象,建立新冠肺炎中药候选活性成分数据库。利用中药系统药理学分析平台数据库(TCMSP)[19],以中药名称为关键词,以口服生物利用度(oral-bioavailability,OB)≥30%和类药性(drug-likeness,DL)≥0.18为参考指标,对其活性成分进行搜索并构建新冠肺炎中药候选活性成分数据库。
1.2 药效团模型的建立及验证
1.2.1 训练集和测试集的搜集
根据文献报道搜集具有人源ACE2抑制活性的小分子化合物,同时在Binding Database数据库(http://www.bindingdb.org/)中检索以人源ACE2为靶点的抑制活性成分,用于训练集和测试集的构建。训练集由6个具有一定活性梯度且结构差异较大的小分子组成(活性梯度为:Ki<100,100≤Ki≤1 000,Ki>1 000),用于生成药效团模型。测试集由30个活性化合物和非活性化合物组成,用于药效团模型的评价及验证。
1.2.2 HipHop药效团模型的建立
药效团是指药物活性分子中对活性起着重要作用的“药效特征元素”及其空间排列形式,可认为是大量活性化合物的共同特征。本研究使用Discovery Studio version 3.5软 件HipHop药效团 构建方法,旨在发现配体小分子化合物的共同特征。按活性排列训练集中小分子化合物,将其中活性较高者Principal设置为2,其他5个活性较低化合物Principal设置为1。所有化合物MaxOmitFeat均设置为1。将正电中心(positive ionizable,PI)、氢键供体(hydrogenbond donor,D)、氢键受体(hydrogen bond acceptor,A)、疏 水 基 团(hydrophobic region,H)、芳环(ringaromatic,R)作为药效团的特征元素进行评价。药效团元素的设定范围为0~5,选择Best模式进行运算,获得10个药效团模型。
1.2.3 药效团模型评价验证
本研究使用Ligand Profiler模块,选择Best模式,用测试集中小分子化合物对药效团模型的性能进行评价,在评分值尽可能高的情况下,选取与活性化合物匹配度较高并与非活性化合物匹配度较低的药效团作为优选药效团模型。
1.3 基于药效团模型虚拟筛选
基于以上得到优选药效团模型,对1.1.2节中构建的新冠肺炎中药候选活性成分数据库进行虚拟筛选。首先,使用Prepare Ligands模块对COVID-19中药候选活性成分数据库中小分子化合物处理,使其转化为3D结构并保存。其次,使用Build 3D Database模块构建3D新冠肺炎中药候选活性成分数据库。最后,使用Search 3D Database模块进行筛选,得到可与人源ACE2结合的潜在SARS-CoV-2抑制活性成分,并使用Filter by Lipinski and Veber Rules模块,遵循Lipinski Rules of Five和Veber Rules原则,通过分子类药性对潜在抑制活性成分进行过滤。
1.4 分子对接
分子对接技术基于配体与受体作用的“锁钥原理”,预测受体与配体的结合模式和亲和力[20]。通过分子对接确定配体小分子与受体蛋白的结合位点和空间位置,有助于研究药物的作用机制和指导新药物的设计[21]。本研究从PDB数据库(http://www.rcsb.org/)下载RBD-ACE2-B0AT1复合体晶体结构(PDB ID:6M17)[22],截取ACE2的一条B链,删去水分子,添加氢原子,即为分子对接的受体。将1.3节中类药性筛选后的小分子化合物作为配体,并进行能量优化、生成三维坐标、添加氢原子等处理[23]。使用LibDock分子对接方式,并选 择Conformation Method为Fast、Docking Preferences为High Quality。根据对接结果初步评价配体与受体的结合情况。为了验证分子对接结果合理性,本文同时使用Auto Dock Vina软件[24]进行配体小分子与ACE2受体的分子对接。采用Auto Dock Vina分子对接方式,对接盒子大小设为88Å×88Å×82Å,能覆盖所有对接位点。对接过程完全采用半柔性对接,ACE2被视为一个刚体,只有配体小分子的构象是可以变化的。
1.5 分子动力学模拟
采用NAMD 2.12软件[25]对小分子化合物与ACE2的最佳对接模型进行进一步模拟和优化。本文运用Discovery Studio version 3.5软件中的Macromolecules模块对ACE2初始结构中缺失的氢原子在中性条件下进行了添加。受体蛋白ACE2采用经典分子力场Amber 03力场进行处理。同时,运用Gaussian 09软件包中的B3LYP密度泛函理论(DFT)对配体小分子进行了几何优化,所有分子的几何优化都使用了6-31G(d,p)基组。在高斯优化的基础上,通过R.E.D.对原子电荷分布的拟合得到相应的小分子力场,其他原子参数采用常规Amber力场(Gaff)进行处理。每个体系放入一个TIP3水盒子中,盒子边界与蛋白质之间最小距离设置为10Å。同时,在水盒子中加入0.15 mol/L的钠离子和氯离子使体系保持电中性,尽可能还原真实的实验环境。首先用共轭梯度算法对所有系统进行10 000步的能量最小化,然后将体系逐渐升温到310 K;温度控制在310 K,所有体系在NVT系综条件下进行2 ns平衡;同样在310 K温度和1 atm压强下,截断值设为12,所有体系在NPT系综下进行了200 ns的分子动力学模拟。
1.6 结合自由能计算
本研究通过分子动力学泊松玻尔兹曼-表面积法(molecular mechanics-Poisson Boltzmann surface area,MM-PBSA),采用分子动力学模拟轨迹最后30 ns等间隔提取的300个构象,计算了小分子化合物与ACE2的结合自由能。MM-PBSA方法已在结合自由能计算中得到了广泛应用[26],具体公式如下:
公式中ΔGcomplex、ΔGprotein和ΔGligand分别表示复合物、受体蛋白以及配体的吉布斯自由能,每一项自由都由ΔEMM、ΔGsolvation和-TΔS 3部分组成,分别表示真空中受体-配体结合前后动力学能量的差值、溶剂化自由能和熵罚。其中动力学项ΔEMM可分为范德华相互作用(ΔEvdW)和静电相互作用(ΔEele),溶剂化自由能包括极性溶剂化自由能(ΔGsol-polar)和非极性溶剂化自由能(ΔGsol-nonpolar)。
2 结 果
2.1 中药处方建立“新冠肺炎中药候选活性成分数据库”
检索文献和临床治疗处方,全面搜集抗COVID-19的有效方剂,最终筛选出143个中药处方作为候选处方。对处方中的中药进行名称规范并进行频数统计,取频次≥5的中药材作为研究对象。共搜集到209种药材,其中59种作为研究对象。利用中药系统药理学分析平台数据库(TCMSP)检索和筛选,获得了640个候选中药活性成分,完成COVID-19中药候选活性成分数据库的建立。COVID-19中药候选活性成分筛选流程图见图1。
2.2 HipHop药效团模型建立及验证
采用HipHop方法,以训练集中的6个活性化合物为对象构建出10个药效团模型。训练集中活性化合物详细信息见表S1,HipHop药效团模型构建结果见表1。10个药效团评分值都超过88,同时其匹配数均为6,说明训练集中6个小分子的共同特征比较明显。图2为药效团验证结果,在评分值尽可能高的情况下,选取与活性化合物匹配度较高及与非活性化合物匹配度较低的药效团作为优选药效团模型。根据药效团热图选择优选药效团,暖色调代表药效团对配体响应度较高,冷色调代表药效团对配体响应度较低。本研究选取药效团08为优选药效团模型进行后续实验。
2.3 优选药效团对“新冠肺炎中药候选活性成分数据库”中潜在SARS-CoV-2抑制成分的虚拟筛选
采用2.2节中构建的优选药效团模型,对2.1节中构建的“新冠肺炎中药候选活性成分数据库”中640个小分子化合物进行虚拟筛选,匹配出107个化合物。再通过“成药五原则”(分子质量<500 u,氢键供体数<5,氢键受体数<10,脂水分配系数<5,可旋转键≤10)对小分子化合物进行类药性筛选,最终得到72个可与ACE2结合的潜在SARS-CoV-2抑制活性小分子。优选药效团08主要是由3个氢键受体(A)、2个疏水中心(H)和1个芳环中心(R)组成(图3a)。图3b-h为优选药效团匹配到的候选成分。表2为虚拟筛选出的部分排名靠前的主要活性成分。
2.4 潜在活性成分与ACE2关键位点的分子对接
为了进一步筛选可以与SARS-CoV-2 S蛋白竞争性结合在ACE2关键活性位点的抑制剂分子,本项研究以2.3节中虚拟筛选出的72个潜在抑制剂小分子为配体,以人源ACE2蛋白为受体进行了分子对接研究。宋磊等[27]通过点突变实验发现SARSCoV-2 S蛋 白 的GLY502、TYR505、TYR489、LYS417、ASN487、ASN501在与ACE2分子识别时起到至关重要的作用。本文通过反向找靶确定了ACE2中与上述残基具有关键相互作用的氨基酸残
基(GLN24、THR27、TYR83、TYR41、LYS353、LYS354),并以此为对接活性位点。采用Libdock半柔性对接方式进行分子对接。每种小分子化合物对接结果中出现多种结合构象,在打分值尽可能高的情况下,选择S蛋白RBD与ACE2的结合位点处关键氨基酸残基呈现较强相互作用的小分子化合物为潜在活性成分。如表3所示,共筛选出10个小分子化合物,10个小分子的详细信息见表S2。为了验证对接结果合理性,同时使用Auto Dock Vina软件验证分子对接结果。在Libdock筛选出10个化合物中,有3个化合物与S蛋白/ACE2识别位点处关键氨基酸残基具有较强相互作用(图4)。
2.5 分子动力学模拟验证
分子对接无法充分考虑蛋白质结构的柔性,为了进一步阐明候选药物与受体蛋白的关键相互作用,以及候选药物的结合对S蛋白与ACE2识别的影响,本项研究对最佳分子对接模型进行了分子动力学模拟。在200 ns的分子动力学模拟中,3个化合物与ACE2的均方根偏差(RMSD)都达到了相对平衡的状态(图5,图S1)。MM/PBSA方法计算得到的结合自由能收敛(图S2)。如表4所示,3个小分子化合物与ACE2的结合自由能均为负值,MOL012556具有最好的结合自由能((-14.8±0.5)kcal/mol),其 次 为MOL003306((-14.6±0.4)kcal/mol),MOL004866的 结 合 自 由 能 最 弱((-10.5±0.5)kcal/mol)。此外,从结合自由能分解项可以看出候选化合物与ACE2的结合主要是由范德华相互作用力(ΔEvdW)驱动的。
经过200 ns的分子动力学模拟,最终只有MOL012556小分子依然处在最初结合位置,其他两个小分子结合位点发生改变,移动到ACE2腔体深处。图6展示了MOL012556、MOL004866、MOL003306三个小分子与ACE2在结合自由能最低时候的代表性结合方式与相互作用。对于MOL012556(图6a),在结合位点处,其C21、C39和C15位羟基分别与ACE2的ASP350(2.6Å)、MET383(2.7Å)、ALA386(3.0Å)形成3个氢键相互作用,LYS353、ARG393与MOL012556通过静电相互作用结合在一起。除此之外,GLY354、PHE356和ALA387与MOL012556通过疏水相互作用结合在一起。MOL003306与MOL012556具有相似的结合自由能,但表现出不同的结合方式。MOL003306主要通过疏水相互作用结合在ACE2腔体深处,其中包括:PHE40、TRP69和PHE390与配体间的Pi-Pi疏水堆积作用;LEU73、LEU391与配体间的Pi-Alkyl疏水作用;以及GLN102、ASP350与配体间的静电相互作用(图6c)。MOL004866(图6b)、C18位 或C19位 羟 基 与ASP382通过氢键相互作用结合。此外,MOL004866通过静电相互作用与ASP350、ASP382和LYS562结合,通过疏水相互作用与PHE40、PHE390和LEU391结合。
因此,上述3个候选药物都可以与ACE2形成稳定的复合物,尤其是MOL012556可以结合在S蛋白与ACE2的识别位点。图7为MOL012556结合前后S蛋白与ACE2的结合模型图,黄色卡通呈现的是S蛋白,灰色卡通为无配体时的ACE2受体,玫红色卡通为加入MOL012556小分子后ACE2构象。从动力学模拟结果看,当无抑制剂分子时(图7a),ACE2氨基酸残基TYR41的侧链乙基-苯酚羟基与S蛋白氨基酸残基THR550上的主链氧原子形成氢键相互作用。此外,ACE2的氨基酸残基LYS353分别与S蛋白氨基酸残基GLY496、ASN501、GLY502形成较强的氢键相互作用。然而,当MOL012556结合以后(图7b),S蛋白与ACE2间的关键相互作用,尤其是上述氢键相互作用完全丧失。整体来看,MOL012556小分子与ACE2结合方式和相互作用更优,两者的结合能有效阻断SARS-CoV-2 S蛋白与宿主细胞ACE2结合,进而阻断病毒感染人体。综上,本文推测MOL012556小分子极有可能是一种潜在的SARSCoV-2抑制剂。
3 讨 论
为了探索对SARS-CoV-2具有潜在抑制作用的中药活性成分,本文首先检索了COVID-19治疗中药处方143个,代表处方有荆防败毒散、柴胡达胸合剂、疏风解毒胶囊、连花清瘟胶囊、清肺达原颗粒、化湿败毒方、宣肺败毒方、止咳枇杷合剂等。其中代表性药材包括黄芪、黄芩、连翘、柴胡、甘草、青蒿、枇杷叶等,这些药材被报道在抗SARSCoV-2中起到一定作用[28],表明本文取样和统计方法的可靠性。本文进一步采用基于配体的虚拟药物筛选,分子对接和分子动力学模拟等方法探究了这些药材中的活性成分与ACE2蛋白的互作机制。结果表明,MOL012556小分子与ACE2的结合能有效阻断SARS-CoV-2 S蛋白与宿主细胞ACE2识别的关键相互作用。因此,推测MOL012556小分子为一种潜在的SARS-CoV-2抑制剂。
MOL012556小分子全称为23-trans-pcoumaryhormentic acid,其OB为36.08,DL为0.32。23-trans-p-coumaryhormentic acid是中药枇杷叶中有效活性成分,代表处方有五叶芦根汤代茶饮、新冠康复颗粒、薛氏五叶芦根汤、银连解毒汤、止咳枇杷合剂。枇杷叶主要含有黄酮类、三萜酸类、有机酸类、挥发油类等化学成分,主要用于清肺止咳、降逆止呕,同时还具有抗炎、祛痰、止咳、抗肺纤维化、抗氧化、降血糖、抗肿瘤、止呕等药理作用[29]。向阳等[30]基于网络药理学与分子对接法探讨薛氏五叶芦根汤防控COVID-19的作用机理,发现薛氏五叶芦根汤中多种活性成分可通过与COVID-19的相关靶标结合,通过多种生物学途径对COVID-19发挥调控作用。周瑞等[31]预测止咳枇杷合剂干预COVID-19的作用机制得出结论:枇杷叶作为方剂中主要中药,其活性化合物可能通过抗炎、抗病毒复制等过程发挥多靶点、多通路、多成分干预COVID-19的作用。由此可见,枇杷叶抗炎、祛痰、止咳、抗肺纤维化等疗效在COVID-19治疗中发挥巨大作用,其有效活性成分在研发COVID-19特异性抗病毒药物中具有极大的潜力。
目前,已有阻断ACE2和病毒S蛋白相互作用的潜在COVID-19药物的研究。有研究报道了两种影响ACE2和病毒S蛋白相互作用的小分子,分别为SSAA09E2(-8.04 kcal/mol) 和Nilotinib(-8.34 kcal/mol)[32]。从 亲 和 力 看23-trans-pcoumaryhormentic acid((-14.8±0.5)kcal/mol)优于上述两种分子。SSAA09E2的结合使得ACE2和S蛋白之间产生了更多的氢键和更强的相互作用,尽管该研究中通过动力学分析强调SSAA09E2的结合可对ACE2和S蛋白的结合过程产生影响,但SSAA09E2更有可能是一种ACE2与S蛋白结合的激 动 剂。与SSAA09E2相 比,23-trans-pcoumaryhormentic acid有效阻断了ACE2和S蛋白之间包括氢键在内的相互作用,更有可能开发为SARS-CoV-2抑制剂分子。另外一种分子Nilotinib的结合可通过阻断ACE2和S蛋白之间氢键相互作用进而影响ACE2和S蛋白的结合。23-trans-pcoumaryhormentic acid与Nilotinib有 相 似的 抑 制机制,但不同于Nilotinib(酪氨酸蛋白激酶(abl-tk)抑制剂),23-trans-p-coumaryhormentic acid是已用于COVID-19治疗的中药处方中的活性成分,更有可能开发为SARS-CoV-2抑制剂分子。遗憾的是,限于目前难以获取23-trans-p-coumaryhormentic acid的纯品,本研究还不能从生物实验上验证其对SARS-CoV-2的抑制作用。进一步的实验验证非常值得期待。
4 结 论
本文通过中药处方挖掘和分子动力学模拟,从143个COVID-19治疗中药处方中筛选出10种可与SARS-CoV-2 S蛋白/人源ACE2识别位点结合的中药成分。其中,枇杷叶主要活性成分23-trans-pcoumaryhormentic acid与ACE2具有最高的亲和力,且23-trans-p-coumaryhormentic acid的结合可有效阻断SARS-CoV-2 S蛋白与宿主细胞ACE2的结合,进而阻止SARS-CoV-2感染人体。因此,本文推测23-trans-p-coumaryhormentic acid小分子是一种潜在的SARS-CoV-2抑制剂,同时从原子水平预测了其抑制SARS-CoV-2 S蛋白与ACE2结合的内在机制,这将为SARS-CoV-2特异性抗病毒药物的研发提供新的思路。
附件见本文网络版(http://www.pibb.ac.cn或http://www.cnki.net):
PIBB_20220224_Table S1.pdf
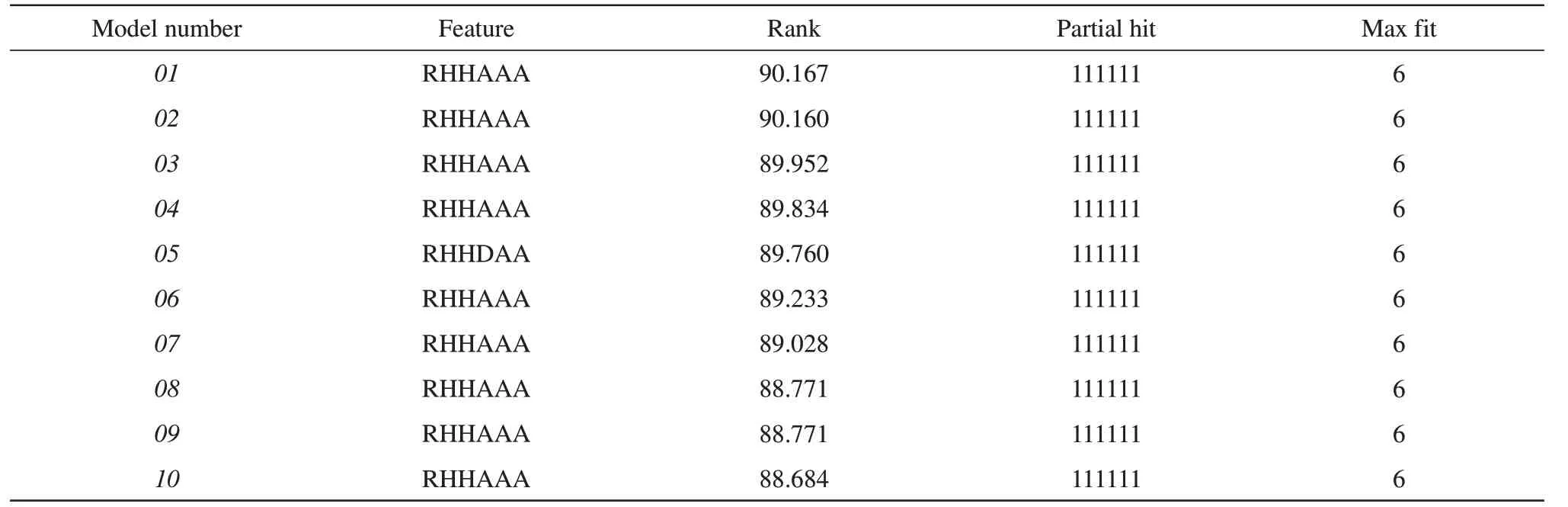
Table 1 Parameters of 10 pharmacophores based on common molecular characteristics

Table 2 Information graph of main active ingredients screened by the virtual screen
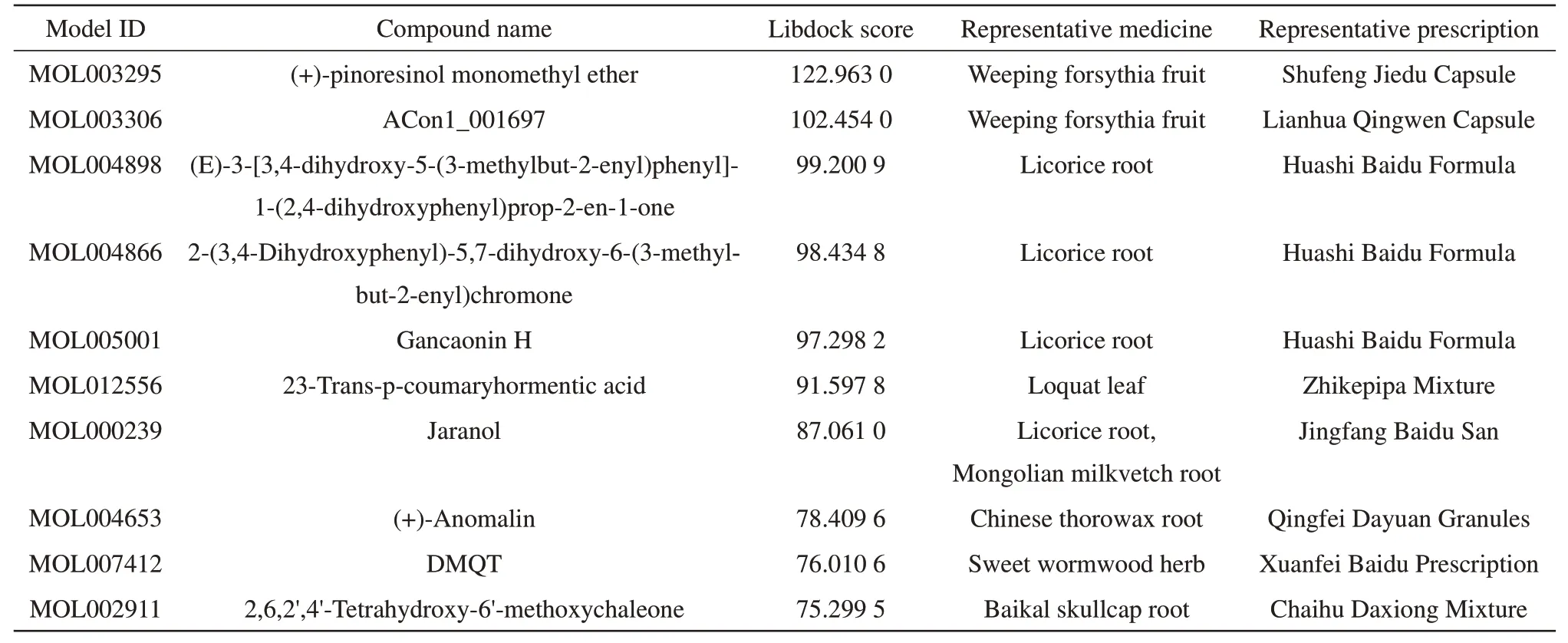
Table 3 Molecular docking results
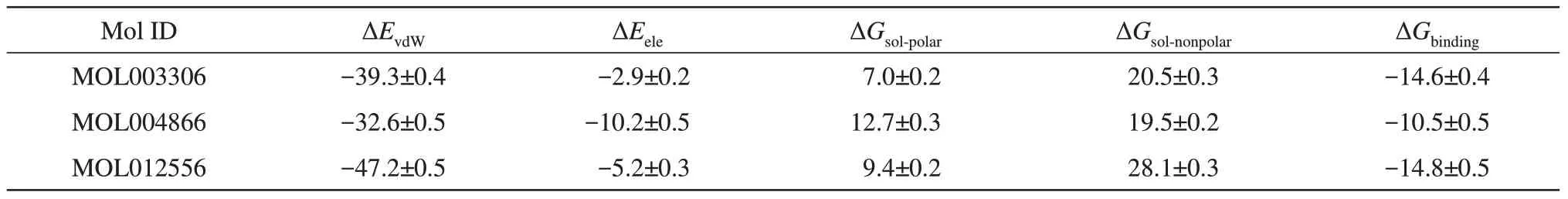
Table 4 MM/PBSA derived binding free energies(kcal·mol-1)of small molecule compounds and ACE2
PIBB_20220224_Table S2.pdf
PIBB_20220224_Fig S1.jpg
PIBB_20220224_Fig S2.jpg
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
小雪花·初中高分作文(2021年4期)2021-08-27
渤海大学学报(自然科学版)(2020年3期)2020-02-02
故事大王(2017年11期)2018-01-21
中学化学(2017年6期)2017-10-16
中学化学(2017年6期)2017-10-16
中学化学(2017年2期)2017-04-01
试题与研究·高考理综化学(2016年3期)2017-03-28
科技创新导报(2016年30期)2017-03-15
