
Transition metal anchored on C9N4 as a single-atom catalyst for CO2 hydrogenation: A first-principles study
2022-10-26 09:47JiaLiangChen陈嘉亮HuiJiaHu胡慧佳andShiHaoWei韦世豪
Chinese Physics B 2022年10期
Jia-Liang Chen(陈嘉亮) Hui-Jia Hu(胡慧佳) and Shi-Hao Wei(韦世豪)
1Department of Microelectronic Science and Engineering,School of Physical Science and Technology,Ningbo University,Ningbo 315211,China
2Department of Electronic and Information Engineering,School of Electrical Engineering and Computer Science,Ningbo University,Ningbo 315211,China
Keywords: first-principles calculation, CO2 hydrogenation, catalysts, electronic structure, reaction mechanisms,reaction paths
1. Introduction
How to effectively alleviate the greenhouse effect has always been a source of concern around the world. With the rapid development of industry and society, human activities such as automobile exhaust and fossil fuel consumption are inevitably accompanied by a large amount of carbon dioxide (CO2) emissions.[1–3]As we all know, the increase of CO2emissions is the main factor of the global greenhouse effect.[4–6]At present,capture,chemical absorption and electrocatalysis are mainly used to reduce the amount of CO2in the atmosphere.[7]Electrocatalysis has attracted much attention because of its low cost, large-scale application and sustainability. Furthermore, in addition to the hydrogenation of CO2,the content of CO2can also be controlled. Other methods,such as photocatalysis and CO2electrolysis,can also produce valuable by-products, e.g., methane, formic acid (FA),methanol, etc.[8–10]FA is widely used in chemistry, industry,medicine and other fields,[11–13]so both experimental research and theoretical calculations are dedicated to finding new catalytic materials to convert CO2into FA.[14–19]Unfortunately,due to the high cost,low catalytic efficiency,and harsh external reaction conditions required for different CO2hydrogenation catalytic materials, it is difficult to achieve under normal temperature conditions.As a result,finding a suitable catalytic material for CO2hydrogenation to formic acid is still a very valuable research.
Since the single-layer graphene is successfully separated,[20,21]carbon-based materials have been widely studied as a substrate with excellent catalytic effects in substances conversion.[22–30]Obviously, because of excellent properties, such as inherent thermal stability, semiconductivity, and cheapness, great efforts have been made to find a carbon-based catalyst that can convert CO2into FA.[31–36]However,considering that carbon itself is inert to certain electrochemical reactions,pyrolysis is usually used in experiments to prepare transition metal (TM) and nitrogen-doped carbonbased materials.[43,44]Some carbon atoms in the carbon-based materials are replaced by nitrogen atoms to form carbon nitrogen (CN) materials, which not only retains the original stability, but also provides ideal sites for metal atoms due to the electron pairs of nitrogen atoms. For example, the high throughput single photon excitation pathways, photocatalytic hydrogen and oxygen release, and Au deposition over WO3/g-C3N4Z-scheme heterojunction have all been investigated.[39–41]Furthermore, the doping-induced method significantly improves the photocatalytic activity and organic matter degradation ability of g-C3N4.[42,43]In experiments,common methods, such as chemical vapor deposition, alkaline activation and thermal stripping method, wet chemical method and salt fusion synthesis, are utilized to generate CN materials.[44–46]In addition, since Pt1@FeOxis firstly proposed to be used in electrocatalysis,[47]single-atom catalysts(SACs) have become the research frontier. Similarly, there are many methods for preparing SACs of CN materials. For example, Co-NG sheet is generated by sonication and heat treatment,and Fe/N/graphene monolayer are prepared by ball milling,demonstrating that CN materials can be employed as substrate for SACs.[48,49]Many studies have shown that SACs formed by doping CN materials with metal atoms have better reactivity and selectivity.[50–58]For instance, Yuanet al. reported that,compared with pure CN samples,doping Ni atoms in N-doped carbon nanosheets can significantly improve the CO2conversion performance.[59]Maet al.reported that Cu doping in C2N has good stability and satisfactory performance(the highest barrier is only 0.53 eV)of CO2hydrogenation into FA using density functional theory (DFT).[60]Liet al.analyzed and summarized the catalytic effects of SACs formed by different 2D single-layer nanosheet doped with Cr,Mn,Fe,Co,Ni,Cu,and Zn on CO2hydrogenation from experimental and theoretical perspectives, indicating that SACs have exhibited comparable catalytic performance to the noble-metal benchmarks.[61]So far, although SACs have shown great potential in CO2hydrogenation,the research on CO2hydrogenation is still in its infancy and needs further exploration.
The above studies fully show that SACs formed by TMdoped CN material are promising candidate for hydrogenation of CO2to form FA. Therefore, it is of great significance to find a suitable new CN material as the TM atom carrier. Fortunately, Niu’s group just predicted a new type of CN material, C9N4.[62]The calculation results show that, compared with other CN materials synthesized in the experiment(such as C2N, g-C3N4), C9N4formation energy is lower,[63–65]which means that it is very possible to experimentally synthesis C9N4and generate SACs utilizing current experimental methods.Therefore,we have carried out the research on the hydrogenation of CO2to FA catalyzed by TM doped C9N4and explained its catalytic mechanism, hoping to provide a little bit of help for the experimental synthesis SACs based on C9N4.
In order to find a good catalyst for CO2hydrogenation to formic acid and clarify its corresponding catalytic mechanism,we study the CO2hydrogenation performance of sixteen different TM (TM = Cr, Mn, Fe, Co, Ni, Cu, Zn, Ru, Rh, Pd,Ag,Cd,Ir,Pt,Au,and Hg)atom doped into the C9N4monolayer. Our research results show that Cu@C9N4has the best catalytic performance,and its maximum energy barrier is only about 0.41 eV. In addition, We also find three other systems(Co@C9N4,Ir@C9N4and Ni@C9N4)also have excellent catalytic effects in the hydrogenation of CO2,and their maximum energy barriers are all less than 0.50 eV, which is lower than that of Maet al.[60]All of the above shows that C9N4is a new type of realizable 2D CN material with promising prospects.
2. Computation details
In our work, all chemical spin-polarized density functional theory (DFT) calculations are performed by using the projector augmented wave (PAW) potentials as implemented in the Viennaab initiosimulation package (VASP).[66–68]And the generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation energy density functionals is adopted.[69,70]To describe the electron-exchange correction effect, van der Waals (vdW) of DFT-D3 method is adopted.[71]The plane wave basis sets with a kinetic energy cutoff of 500 eV are used. The 3×3×1 Monkhorst–Pack mesh is used in sampling the Brillouin zone.A vacuum space of 20 ˚A is adopted to avoid the interactions between adjacent layers. The energy and force of structural geometry optimization are accurate to 10-5eV and 10-2eV,respectively. Isobaric–isothernal (NPT) ensemble molecular dynamics (MD) simulations are carried out at 300 K and 1000 K with an integration step of 1 fs. The diffusion and reaction of the system are invested along the minimum energy path(MEP)with the climbing image nudged elastic band(CINEB)method.[72]
The adsorption energy(Eads)is defined as

whereEbulkandEsingle-atomrepresent the energy of TM crystal and a single TM atom, respectively. Andnis the number of TM atoms in the TM crystal.
3. Result and discussion
3.1. Stability of TM@C9N4
After optimized,the lattice constant of the C9N4unit cell isa=b=9.64 ˚A,which contains 18 carbon atoms and 8 nitrogen atoms. If the radius of N atom is not considered here,the diameter of the hole formed by six nitrogen atoms is about 5.51 ˚A(see Fig.1(a)),coinciding with previous reports.[74,74]Because of the large size of the hole,it provides many chemically active sites. The nitrogen atoms in the hole have lone pair electrons,which is the reason why TM atom can be firmly combined with the substrate. As shown in Fig. 1(a), thirteen possible anchoring sites are taken into consideration: S1–S13,and the blue and orange dots indicate that adsorption sites are on the same plane as C9N4and adsorption sites are above C9N4,respectively. All metal atoms are investigated on these sites,and finally three different most stable adsorption modes are found, as shown in Fig. 2. It is found that Cr, Mn, Fe,Co, Ni, Cu, Zn, Ru, Rh, Pd, Cd, Ir, and Pt tend to bond with two nitrogen atoms and lie in the same plane with the substrate(Fig. 2(a)). At the same time, Ag, Au and Hg are located in the center of the hole (Figs. 2(b) and 2(c)). The difference is that Au or Ag is coplanar with C9N4monolayer, while Hg is above the C9N4monolayer (the vertical distance between Hg and C9N4monolayer is about 1.06 ˚A). The adsorption energy between TM atom and C9N4monolayer is shown in Fig. S1 and Table S1. The large adsorption energy (all great that 2.0 eV)indicates the formation of a strong chemical bond(except for Hg). The stability of SACs is further confirmed by MD simulations. We calculate the stability of six TM@C9N4(TM = Co, Ni, Cu, Ru, Rh, and Ir) and find that they all have satisfactory stability. For example,as shown in Figs.S2 and S3,the structures of Cu@C9N4has no obvious change at 300 K and 1000 K,and the energy fluctuation is minor. In addition, we also calculate the partial density of states (PDOS)of TM@C9N4(Fig. S4). It is found that there is a strong orbitals hybridization between the d orbitals of TM atom and the p orbitals of the nitrogen atoms near the Fermi level. These results further confirm that TM atom can be firmly anchored to the hole of C9N4. This conclusion is also confirmed by the charge transfer between TM atom and C9N4and the diffusion barrier of TM atom on C9N4. For simplicity, here, Cu atom is adsorbed on C9N4as an example. Obviously, as shown in Fig.1(b),the charge is transferred from Cu atom to N atoms.The Bader charge analysis shows that about 0.75 electrons are transferred from Cu atom to C9N4, where the single N atom connected to Cu atom gets about 0.31 electrons. We also find that, as shown in Fig. S5, if the adsorbed Cu atom leaves the adsorption position, it needs to overcome the energy of 3.24 eV or 3.16 eV,which indicates that the adsorbed Cu atom is difficult to move from the hole to the adjacent hexagonal adsorption position composed of C and N atoms. In addition,as shown in Table S1,a ratio of adsorption energy to cohesive energy is greater than 0.5, which means that TM atoms are not easy to form clusters during the reaction and influencing the activity of the catalyst.[75,76]Therefore,TM@C9N4can be regarded as a stable CO2hydrogenation catalyst.

Fig.1. (a)S1–S13 represent the adsorption position of adsorbed metal atom on C9N4,where the solid parallelogram represents the C9N4 unit cell, and (b) the difference charge density of Cu atom adsorbed on C9N4, where the isosurface value is set to be 0.003 e/˚A3, and the blue and red isosurfaces represent the accumulation and consumption of electrons,respectively.
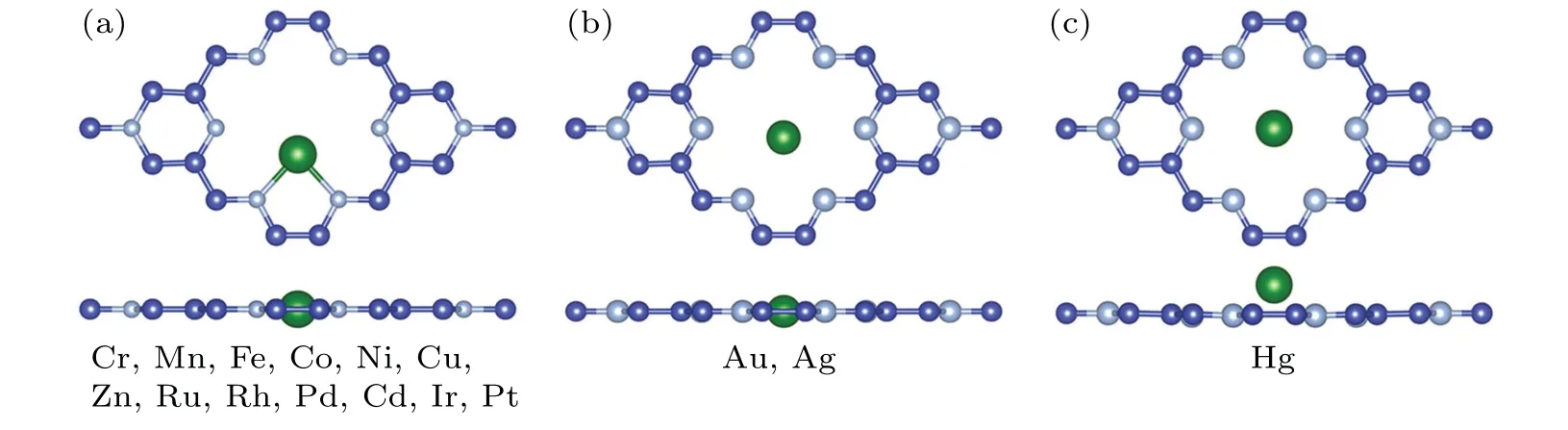
Fig.2. (a)–(c)The most stable state of the adsorption state of sixteen metals.
3.2. Adsorption of H2 and CO2 molecules on TM@C9N4


Table 1. The adsorption energy(eV)of H2 (),CO2 ()and H2 +CO2 (E)for the most favorable gas adsorption. The bond length(˚A)of H–H for H2 molecule singly adsorbed()and H2+CO2 co-adsorbed()on TM@C9N4. e is the vertical height of CO2 from TM@C9N4 at the co-adsorption state. The red font represents that is greater than .
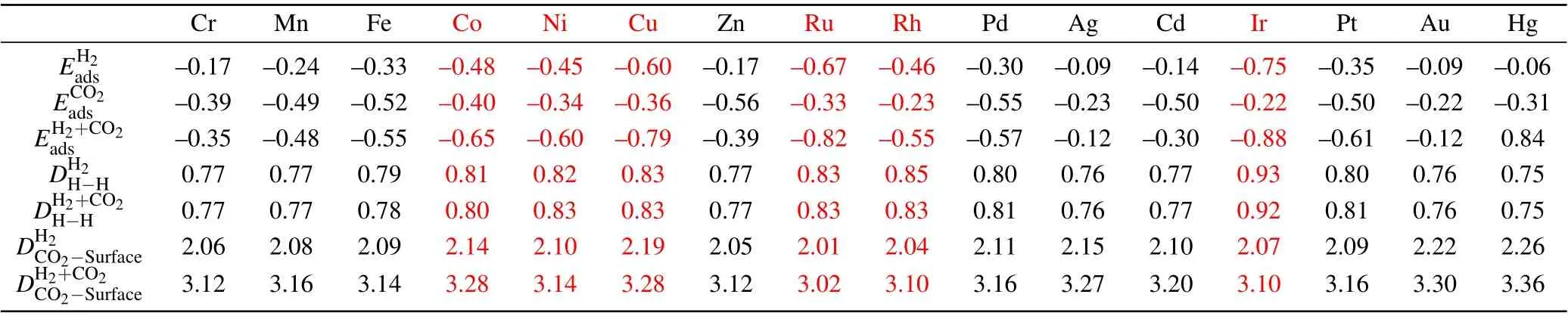
Table 1. The adsorption energy(eV)of H2 (),CO2 ()and H2 +CO2 (E)for the most favorable gas adsorption. The bond length(˚A)of H–H for H2 molecule singly adsorbed()and H2+CO2 co-adsorbed()on TM@C9N4. e is the vertical height of CO2 from TM@C9N4 at the co-adsorption state. The red font represents that is greater than .
Cr Mn Fe Co Ni Cu Zn Ru Rh Pd Ag Cd Ir Pt Au Hg EH2 ads –0.17 –0.24 –0.33 –0.48 –0.45 –0.60 –0.17 –0.67 –0.46 –0.30 –0.09 –0.14 –0.75 –0.35 –0.09 –0.06 ECO2 ads –0.39 –0.49 –0.52 –0.40 –0.34 –0.36 –0.56 –0.33 –0.23 –0.55 –0.23 –0.50 –0.22 –0.50 –0.22 –0.31 EH2+CO2ads –0.35 –0.48 –0.55 –0.65 –0.60 –0.79 –0.39 –0.82 –0.55 –0.57 –0.12 –0.30 –0.88 –0.61 –0.12 0.84 DH2 H-H 0.77 0.77 0.79 0.81 0.82 0.83 0.77 0.83 0.85 0.80 0.76 0.77 0.93 0.80 0.76 0.75 DH2+CO2 H-H 0.77 0.77 0.78 0.80 0.83 0.83 0.77 0.83 0.83 0.81 0.76 0.77 0.92 0.81 0.76 0.75 DH2 CO2-Surface 2.06 2.08 2.09 2.14 2.10 2.19 2.05 2.01 2.04 2.11 2.15 2.10 2.07 2.09 2.22 2.26 DH2+CO2 CO2-Surface 3.12 3.16 3.14 3.28 3.14 3.28 3.12 3.02 3.10 3.16 3.27 3.20 3.10 3.16 3.30 3.36

Fig.3. The optimized geometric structures for TM@C9N4 (TM=Co,Ni,Cu,Ru,Rh,Ir)adsorbing(a)H2,(b)CO2,and(c)H2+CO2.


3.3. CO2 hydrogenation on TM@C9N4
According to different adsorption conditions,the adsorption of H2alone (named Path I) and the co-adsorption of H2and CO2(named Path II) on TM@C9N4(TM = Co, Ni, Cu,Ru,Rh and Ir)are studied as follows:

The structure of the initial state (IS), intermediate state(MS),transition state(TS)and final state(FS)and the energy change along the MEP are shown in Figs. 4 (for Path I), S7(for Path II)and Table S2. The energy fluctuation diagram of CO2hydrogenation on Cu@C9N4and the number of images of CI-NEB calculated at each step are shown in Fig. S8. As shown in Fig. S7 and Table S2, considering that the energy barriers of the rate-limiting step (RLS) in Path II are greater than 1.60 eV,this means that the reaction along the Path II is difficult to occur at room temperature. For the sake of simplicity,we take Cu@C9N4in Path I as an example in the following discussion. First of all, in the IS, a H2molecule is adsorbed on Cu@C9N4. According to the previous analysis,the H–H bond is activated to a certain extent after the adsorption, so the fracture of H–H bond in the first TS (TS1) only needs to overcome 0.21 eV energy barrier and reach the first MS (MS1). In the MS1, one of the H atoms in H2molecule still forms a bond with the Cu atom (the bond length of H–Cu is about 1.51 ˚A), the other H atom approaches to the N atom in the six nitrogen hole, forming a N–H bond(1.04 ˚A),at the same time, the bond length of H–H is extended from 0.83 ˚A to 0.99 ˚A. For the MS1+CO2state, a CO2molecule is adsorbed from outside. Next, in the second TS (TS2), the Cu–H bond length extends from 1.51 ˚A to 1.61 ˚A, and the energy barrier of 0.41 eV needs to be overcome when CO2molecule approaches the catalyst. After reaching the highest point of energy,CO2spontaneously forms bond with Cu atom and releases energy,forming Cu–O bond(1.87 ˚A).Cu–H bond is broken to form H–C bond (1.12 ˚A). Then in the third TS(TS3), the N–H bond (1.06 ˚A) is broken and the H atom approaches to the O atom, forming a H–O bond (1.07 ˚A), and then forming a FA on the surface of the catalyst. In the whole electrocatalysis process,the highest energy barrier is 0.41 eV.So,the hydrogenation of CO2to FA through the Path I is relatively easy to achieve at room temperature,which is consistent with the results of Kathalikkattilet al.[77]In addition,we also notice that the highest energy barrier of Co@C9N4,Ir@C9N4and Ni@C9N4are 0.43 eV,0.46 eV and 0.48 eV,respectively,which are less than 0.5 eV. While the highest energy barrier of Rh@C9N4is only about 0.51 eV.Just like Cu@C9N4,their RLS is in MS1+CO2→TS2.
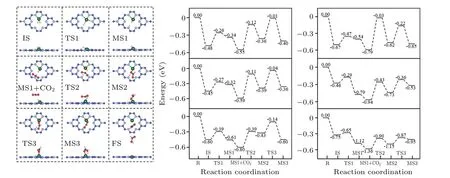
Fig.4. The structure of the intermediate and transition state of Cu@C9N4 (left)and the potential energy profile of TM@C9N4 (TM=Co,Ni,Cu,Ru,Rh,Ir)(right)in Path I.
In addition, we also consider other possible reduction reactions, such as the hydrogenation of CO2to methane or methanol. But in fact, whether it is to generate methane or methanol,it must first reduce CO2to CO,namely,CO2+H2→CO+H2O.Taking MS1+CO2in Path I as the initial state(IS),the corresponding structure and energy change along the MEP for CO2reduction are shown in Fig. S9. It can be seen that there are two large energy barriers in the whole process,which are 1.38 eV and 0.71 eV, respectively. Therefore, the reduction of CO2to methanol or methane is less competitive than the hydrogenation of CO2to formic acid.
4. Property analysis
As mentioned above, considering that Cu@C9N4and Co@C9N4have excellent catalytic performance, the partial density of states (PDOS), projected crystal orbital Hamilton population (pCOHP) and charge transfer between adsorbed gas molecule(H2or CO2)and catalysts for TM@C9N4(TM=Co,Ni,Cu,Ru,Rh,and Ir)are performed to further clarify the catalytic mechanism. Figure 5 shows the PDOS and pCOHP of the transition state TS2 of Co@C9N4(a) and Cu@C9N4(b)in Path I,and Co@C9N4(c)and Cu@C9N4(d)in Path II,respectively. It is obvious find that, from Figs.5(a)and 5(b),the 3d orbitals of Cu or Co atom are strongly hybridized with the molecular orbitals of CO2near the Fermi level in Path I.At the same time,in Path II,near the Fermi level,the hybridization between 3d orbitals of Cu or Co atom and CO2molecular orbitals are much weaker, which can be clearly seen from Figs. 5(c) and 5(d). We can further confirm this conclusion from pCOHP.As shown in Fig.5,the hybridized energy levels of TM-3d orbitals and CO2orbitals split into the bonding and anti-bonding states. The anti-bonding states for Co@C9N4(a)and Cu@C9N4(b)in Path I are all above the Fermi level,while the anti-bonding states of Co@C9N4(c)and Cu@C9N4(d)in Path II are partly under Fermi level. In other words, the interaction between CO2and TM@C9N4gets weakened by the anti-bonding population entering below the Fermi level. This means that the activation of CO2in Path I is stronger than that in Path II. So the energy barriers in Path I are much smaller than those in Path II.

Fig. 5. The partial density of states (PDOS) and projected crystal orbital Hamilton population (pCOHP) for the transition state TS2 of Co@C9N4 (a)and Cu@C9N4 (b)in Path I,and Co@C9N4 (c)and Cu@C9N4 (d)in Path II,respectively. The Fermi level is set to 0 eV.
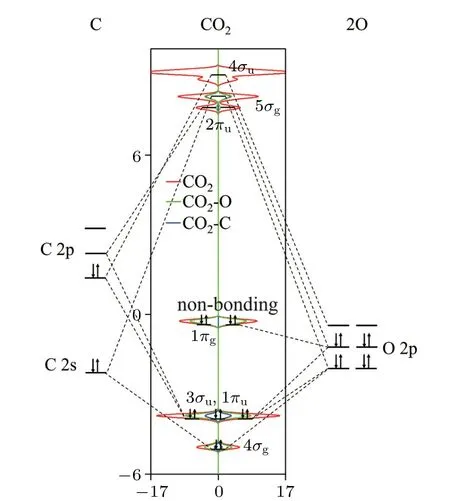
Fig. 6. The calculated PDOS of CO2 and the schematic molecule orbitals energy level diagram of the interaction between C and O atoms.The Fermi level is set to 0 eV.
It is worth noting that the RLS in the catalytic reactions of all systems is MS1+CO2→TS2(shown in Fig.4),which is due to the fact that CO2molecule has a special 1πgorbitals,that is,non-bonding orbitals. As shown in Fig.6,the molecular orbitals of free CO2molecule,mainly including 4σg,3σu,1πu,1πg,2πu,5σgand 4σu.1πgand 2πuorbitals,which corresponds to the highest occupied orbitals(HOMO)and the lowest unoccupied orbitals (LUMO), respectively, are the main participants in the catalytic activation of CO2. However, as shown in Fig.6, the delocalized 1πgbond does not hybridize with C atom, which is only composed of 2p orbitals of two oxygen atoms,so it is a non-bonding orbitals. Moreover,CO2molecule does not have a lone pair of electrons. So the electrons on HOMO have poor activity,resulting in poor chemical activity of CO2, when CO2reacts with the catalyst. Therefore, the RLS (MS1+CO2→TS2) is the process in which CO2participates in the reaction,and the strength of the interaction between the 1πgorbitals and the catalyst determines the difficulty of this reaction.
The PDOS of TS2 for the TM@C9N4(TM=Co,Ni,Cu,Ru, Rh, and Ir) in Path I are shown in Fig. 7. As shown in Fig.7(c), near the Fermi energy level, there is significant hybridization between the 3d orbitals of Cu atom and the 1πgorbitals of CO2. So,in the process from MS1+CO2to TS2,the C=O bond of CO2is extended from 1.17 ˚A to 1.23 ˚A,which means CO2molecule is activated on the Cu@C9N4.While the H–Cu bond is broken from TS2 to MS2(as shown in Fig.4),resulting that CO2maybe replace the position of H to form a bond with Cu. For Co@C9N4, we also find the same hybridization mechanism from Fig. 7(a). We also notice that,for Ni@C9N4, the hybrid interaction between the 3d orbitals of Ni atom and the 1πgorbitals of CO2is much weaker than that of Cu or Co atom, which leads to a larger energy barrier(0.48 eV).

Fig.7. The partial density of states(PDOS)for the TM-nd(n=3,4,5),C,O and H of the transition state(TS2)in Path I.The black,blue,green and red lines represent the TM-nd, carbon, oxygen and hydrogen atoms, respectively. The vertical dash-dotted lines correspond to the Fermi level (set to 0 eV).For the convenience of comparison,the PDOS of Co@C9N4 is plotted twice.
We notice that Co, Rh and Ir are also the same group of TM atoms in the periodic table of elements, and the hybridization of the 3d orbitals of Co atom with the 1πgorbitals of CO2molecule are more obvious than Rh and Ir, while the hybridization of H and Co is relatively weak. The result indicates that the rupture of the Co–H bond and CO2activation are less obstructive for Co@C9N4. So the RLS barrier of Co@C9N4(0.43 eV)is lower than that of Rh@C9N4(0.46 eV)and Ir@C9N4(0.51 eV).The hybridization between CO2and Rh, Ir atoms is also similar. As shown in Figs. 7(e)and 7(f),the hybrid interaction between 5d orbitals of Ir atom and 1πgorbitals of CO2is slightly stronger than that of Rh atom,which leads to that the energy barrier of Ir@C9N4(0.46 eV)is lower than that of Rh@C9N4(0.51 eV). Finally, in the PDOS of Ru and Rh (see Figs. 7(g) and 7(h)) in the same period, the 1πgorbitals of CO2on Ru@C9N4remain narrow and isolated.The weaker interaction between CO2and Ru in the RLS of Ru@C9N4confirms that Ru@C9N4has the worst catalytic performance and the highest energy barrier in these six systems.
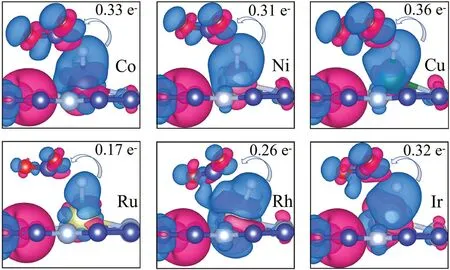
Fig. 8. Difference charge density of CO2 molecule adsorbed on six TM@C9N4 (TM = Co, Ni, Cu, Ru, Rh and Ir) for the transition state TS2 in Path I,respectively.Red and blue represent charge accumulation and depletion,respectively. The isosurfaces is set to be 0.004 e/˚A3.
Additionally, the Bader charge analysis and difference charge density are also performed to calculate the charge transfer between the catalyst and CO2. The charge transfer is observed in Fig.8 from TM atom to CO2. And the relationship between the energy barrier and the number of electrons obtained by CO2from the catalyst is shown in Fig.9. It can be seen that the number of electrons transferred is inversely proportional to the value of the energy barrier that needs to be overcome in the RLS.In other words,the more significant the interaction between CO2and TM@C9N4, the easier it is for the electrons on the d orbitals of the TM atom to be transferred to the CO2molecule to activate CO2.
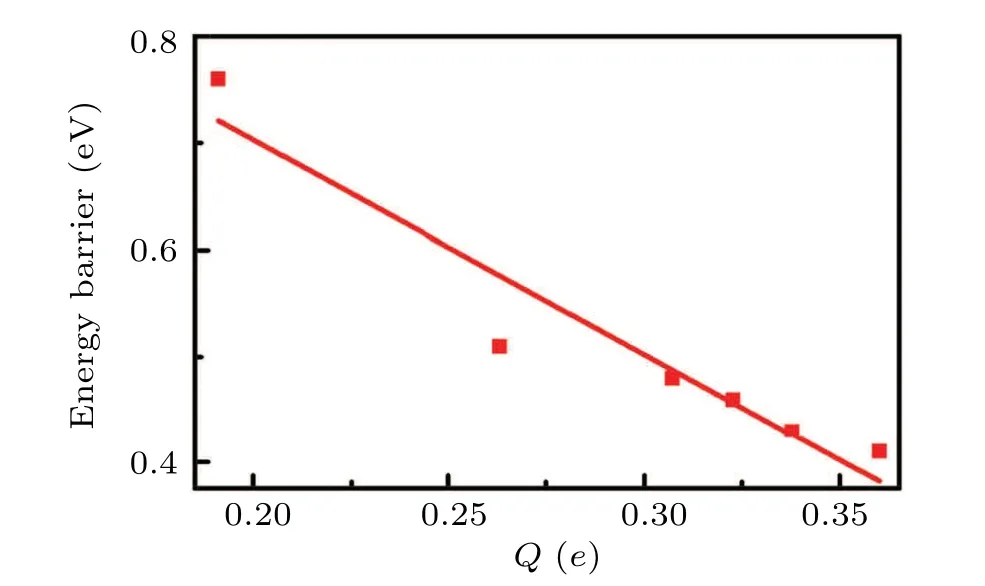
Fig.9. The change of energy barriers as function of the number of electrons(Q)transferred from catalyst to CO2 for the TS2 in Path I.
Furthermore,we also considered the possibility of the aggregation of two TM atoms in the cavity of C9N4. The optimized structures of the co-adsorption of two TM atoms are shown in Fig.S10. It is interesting to find that two TM atoms are located on two sides of C9N4,respectively. It means that,in the process of CO2hydrogenation, a single TM atom still plays a key role.At the same,the energy barrier of RLS for the co-adsorption of two TM atoms is much larger than that of the single TM atom in the CO2hydrogenation catalytic reactions.
5. Conclusion
In general, based on the first-principles calculation, we have studied the catalytic effect of a series of TM(TM=Cr,Mn, Fe, Co, Ni, Cu, Zn, Ru, Rh, Pd, Ag, Cd, Ir, Pt, Au, and Hg)doped with C9N4on CO2hydrogenation. Because of the abundant chemically active sites with lone pairs provided by the 6N hole,TM can be stably doped on C9N4. The extremely high diffusion barrier of Cu atom (3.21 eV or 3.16 eV), results of MD simulations and the strong interaction between TM atom and C9N4demonstrate the catalytic performance and stability of the catalyst. It is interesting to find that,except for the six TM atoms (Co, Ni, Cu, Ru, Rh and IR), other TM atoms can be poisoned by CO2. We have discussed the two routes(Path I and Path II)of CO2hydrogenation respectively.Our results show that Path I is more in line with our requirements in energy. It is show that among the sixteen TM@C9N4monolayer catalysts, Cu@C9N4and Co@C9N4show better catalytic effects (energy barrier is less than 0.45 eV), which has more advantages over other similar catalysts. The partial density of states(PDOS),projected crystal orbital Hamilton population (pCOHP) and Bader charge analysis for CO2and H2adsorbed on TM@C9N4further demonstrate the catalytic mechanism: (i) When the H2molecule is captured by the catalyst, the hybridization of the s orbitals of H2and the 3d orbitals of the TM atom activates the H–H bond and H2molecule is significantly elongated. (ii) The stronger the hybridization between the non-bonding orbitals of CO2(1πg)and the 3d orbitals of TM atom near the Fermi level, the smaller the energy barrier of the rate-limiting step. That is why the Cu@C9N4system has a minimum energy barrier of 0.41 eV in Path I. In short, our research has discovered a achievable non-noble metal 2D single-layer nanosheet catalytic material,which can alleviate the greenhouse effect and form valuable FA product,providing new possibilities for future sustainable development.
Acknowledgements
Project supported by the National Natural Science Foundation of China (Grant No. 51871126) and the K. C. Wong Magna Fund in Ningbo University. The computation was performed in the Supercomputer Center of NBU.

- Chinese Physics B的其它文章
- Formation of high-density cold molecules via electromagnetic trap
- Dynamics of molecular alignment steered by a few-cycle terahertz laser pulse
- Terahertz spectroscopy and lattice vibrational analysis of pararealgar and orpiment
- Molecule opacity study on low-lying states of CS
- Finite-time Mittag–Leffler synchronization of fractional-order complex-valued memristive neural networks with time delay
- Ultrafast Coulomb explosion imaging of molecules and molecular clusters