
绒山羊皮肤全长转录组测序及生物信息学分析
2022-11-04 09:57苏德其其格AKONYANIZaccheausPazamilala乌兰吐雅吴江鸿
中国农业大学学报 2022年12期
李 颖 宋 峰 苏德其其格 AKONYANI Zaccheaus Pazamilala 乌兰吐雅 吴江鸿
(内蒙古民族大学 动物科技学院,内蒙古 通辽 028000)
内蒙古绒山羊是一种兼具绒肉兼用性的地方优良品种[1],毛被洁白有光泽,是我国重要的创汇动物[2],是经过长期人工选育而成可适应饲养地区寒冷、干旱等气候环境的优良选育能适应恶劣环境变化的绒山羊品种、提高绒山羊优良产绒性状以及环境与基因互作已成为研究热点。既使基因组序列已经公布,但绒山羊参考基因组收录的数据其特征和mRNA 结构并没有得到深入分析且基因信息收录不全,因此不能满足现阶段绒山羊育种及优良性状挖掘等相关研究。伴随着高通量测序技术的迅猛发展,转录组测序(RNA-Seq)技术在各个研究领域得到广泛的推广与运用,成为研究基因表达调控的主要方法[3]。RNA-Seq可用于发现低丰度转录本和新转录本,并识别不同样本之间转录本的差异表达[4-5]。第二代高通量测序技术可用于筛选差异表达基因[6],第三代测序技术可以不间断地直接读取全长转录本,可以有效地获得单个RNA分子的完整高质量序列,并准确识别下一代测序(NGS)无法识别的差异表达异构体、同源基因、超家族基因和等位基因的转录本[7]。它可以改善基因表达的实时定量聚合酶链反应(RT-PCR)结果,识别选择性剪接和基因融合现象,发现新基因和转录异构体[8]。为能够更加全面得到绒山羊皮肤中的基因及转录本,本研究以内蒙古白绒山羊为试验动物,利用 SMRT 技术对绒山羊皮肤进行全长转录组测序及生物信息学分析,通过与绒山羊参考基因组进行数据比对,鉴定新基因及转录本,分析绒山羊皮肤基因的可变剪接(Alternative splicing,AS)类型并统计不同类型的数量,预测绒山羊皮肤中 lncRNA 及简单重复序列(Simple sequence repeats,SSR),为有效提高绒山羊基因组注释水平,丰富基因序列及结构信息,深入挖掘绒山羊品种资源,优良性状的提升及家畜基因组与环境互作机理研究提供理论依据。
1 材料与方法
1.1 试验材料
为更加全面的得到绒山羊皮肤基因的所有转录本,本研究在呼和浩特市土默特左旗分4个时间点(1、4、7和10月)采集了3只内蒙古白绒山羊的体侧皮肤和耳部皮肤样品,使用Trizol分别从皮肤样本中提取RNA,并将样品RNA混合构建文库后进行测序。
1.2 试验方法
1.2.1绒山羊皮肤样本RNA提取
每100 mg新鲜皮肤组织样品中加入1 mL Trizol试剂,使用无菌手术刀在冰上将样品切碎,并用无菌匀浆器匀浆,将组织或细胞裂解液转移到1.5 mL 无RNA酶EP管中。置冰上,静置5 min。每管加入200 μL氯仿,充分混合后冰上静置10 min,使核蛋白复合物完全解离。4 ℃ 13 000 r/min离心15 min。期间,取一新EP管,加入500 μL异丙醇,冰上预冷。离心后,将上层水相 (约500 μL)转移到该新的EP管中。冰上静置,酒精沉淀10 min,13 000 r/min 离心10 min,去除上清液。用1 mL 75%乙醇洗涤RNA沉淀1次,12 000 r/min离心5 min。去掉上清,真空干燥RNA沉淀5~10 min。将RNA溶解在30~50 μL DEPC处理过的去离子水中,最后进行分光光度分析以确定样品浓度和纯度。
1.2.2绒山羊皮肤全长转录组建库及SMRT测序
在文库构建之前,从每个皮肤样本中收集等量的总RNA,根据Iso-Seq协议构建SMART测序文库。使用SmarterTMPCR cDNA synthesis kit合成mRNA的全长cDNA。PacBio Iso-Seq文库按照PacBio标准文库制备方案制备,在PacBio RsII仪器上进行测序,PacBio测序由中国北京百迈客测序公司进行。
1.2.3绒山羊皮肤全长转录组数据处理
使用Iso-seq管道将原始数据进行处理,运行参数为minFullPass=3和minPredictedAccuracy=0.9。然后通过poly A尾部信号和5'及3' cDNA引物来确定全长非嵌合的转录本。ICE(Iterative clustering for error correction)被用来获得共识异构体,使用Quiver对来自ICE的FL共识序列进行抛光,高质量的FL转录本被分类,其标准是校正后的准确率高于99%。使用GMAP将FL共识序列映射到参考基因组上,用Pbtranscript-tofu软件包对映射的读数进行进一步折叠,最小覆盖率(Min-coverage)为85%,最小一致性(Min-identity)为90%,在折叠多余的转录本时不考虑5′差异。
1.2.4绒山羊皮肤全长转录组数据分析
新转录本和新基因功能注释使用BLAST[9]软件与NR、Swissprot、GO、COG、KOG、eggNOG、Pfam、KEGG数据库进行比对,获取注释信息;使用TransDecoder[10]软件对其编码区序列及对应氨基酸序列进行预测;通过Astalavista[11]软件获取每个样品间存在的可变剪接类型;使用Cytoscape[12]插件ClueGO对可变剪接基因进行功能富集分析;利用MEME对所有转录本poly A位点上游50 bp的序列进行分析;从新转录本中筛选500 bp以上的转录本,利用MISA软件做SSR分析;对新发现的转录本进行lncRNA的预测,主要包括:CPC分析、CNCI[13]分析、pfam蛋白结构域分析和CPAT[14]分析4种方法。
2 结果与分析
2.1 绒山羊皮肤SMRT测序数据结果统计
采用SMRT测序技术,完成样品的全长转录组测序,使用2个Cell获得 42.41 Gb clean data的数据。测序获得CCS 591 585 条,其中全长非嵌合序列466 058条。对全长非嵌合序列进行聚类得到一致序列149 604条,对一致序列进行Polish得到高质量一致序列共137 211条,用二代转录组数据对低质量一致序列进行校正,校正后与高质量一致序列合并进行去冗余得到82 382条转录本序列。对去冗余后的82 382个转录本进行可变剪接分析,检测到基因18 919个,其中新基因6 166个,新发现转录本55 875 个。对新发现的转录本进行序列结构分析,共预测出 66 951个SSR、39 346个完整ORF序列和10 927个lncRNA,并完成了49 573个新转录本的功能注释。
2.2 绒山羊皮肤全长转录组测序完整性评估
利用BUSCO[15]对去冗余后的转录组进行完整性评估,BUSCO使用OrthoDB作为参考数据库,构建了多个进化分支的单拷贝基因集。评估结果如图1所示,完整覆盖基因数为3 145个,其中单拷贝基因数1 260个,多拷贝基因数1 885个,占总基因数的79.62%,测序获得的绒山羊皮肤全长转录组数据的完整性及准确性是非常可靠的。
2.3 绒山羊皮肤新转录本及新基因功能注释
将得到的新转录本和新基因与NR、Swiss-prot、GO、COG、KOG、Pfam和KEGG等数据库比对进行功能注释,各数据库注释到的转录本数量统计见表1。由表1可知,共对49 573个转录本进行注释,其中通过NR数据库成功注释的转录本数量最多,共有49 374条,在总注释量中占比99.60%。新转录本NR数据库注释分布前几位的分别是:山羊(Caprahircus,11 873,24.06%)、牛(Bostaurus,9 918,20.10%)、野牦牛(Bosmutus,6 109,12.38%)、绵羊(Ovisaries,4 737,9.60%)和藏羚羊(Pantholopshodgsonii,3 331,6.75%)(图2(a))。新基因NR数据库注释分布前几位的分别是:牛(Bostaurus,1 787,42.65%)、野牦牛(Bosmutus,1 318,31.46%)、山羊(Caprahircus,182,4.34%)、绵羊(Ovisaries,138,3.29%)和野牛(Bisonbison,135,3.22%)(图2(b)),被注释到的物种主要为牛科物种。KEGG分析结果显示:33 200个新转录本被富集到302条通路中,2 252个新基因被富集到245条通路中。GO数据库将43 582个新转录本及3 170个新基因进行了注释,结果可分为三大类,细胞组分、分子功能及生物学过程(图2(c))。一条基因通过内含子的不同剪接可构成不同的转录本,SMRT测序技术可以获得基因详尽的转录本,本研究所预测的转录本数量远高于参考基因组,新转录本及新基因功能注释将完善绒山羊转录组数据信息。
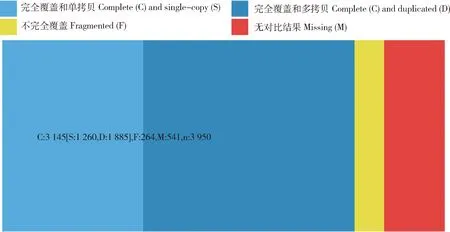
图中n及对应数字为近缘物种单拷贝基因集和该基因集内基因数目。n and corresponding numbers in the figure are single-copy gene sets of closely related species and the number of genes in the gene set.图1 绒山羊皮肤转录组完整性评估分析Fig.1 Analysis of transcriptome integrity assessment in the skin of cashmere goats

表1 绒山羊皮肤新转录本及基因功能注释统计表Table 1 Statistical table of new transcripts and gene function annotations in the skin of cashmere goats
2.4 绒山羊皮肤可变剪接及lncRNA分析
通过Astalavista软件鉴定存在的可变剪接类型,主要的基因可变剪接类型如图3(a)所示。全长转录组测序共鉴定了25 717个可变剪接,其中可变外显子(Mutually exclusive exon)631个、内含子保留(Intron retention)9 899个、外显子跳跃(Exon skipping)9 184个、可变转录起始位点(Alternative 5′splice site)2 325个、可变转录终止位点(Alternative 3′splice site)3 678个(图3(b))。将AS分析得到的新转录本对其编码区序列及对应氨基酸序列进行预测,共获得ORF 49 294条,其中完整ORF 39 346条,预测得到的完整ORF区编码蛋白序列长度分布为:0~100 aa,15 039条(30.51%)、100~200 aa,11 650条(23.63%)、200~300 aa,6 879条(13.96%)、300~400 aa,4 761条(9.66%)及400~500 aa,3 282条(6.66%)。
使用Cytoscape 插件ClueGO对可变剪接基因进行功能富集分析,1 052个可变剪接基因共富集到624个GO term。对富集到的term进行分类发现主要涉及mRNA分解代谢(mRNA catabolic process)、mRNA代谢(mRNA metabolic process)、内皮细胞发育(Endothelial cell development)、肾小球滤过调节(Regulation of glomerular filtration)等生物学过程,其中核糖体蛋白家族和泛素结合酶家族基因是富集到term最多的基因家族。同时找到了一条与皮肤形态发育相关的term(Wnt信号通路,Wnt signaling pathway),并基于可变剪接位点信息发现基因黑素皮质素1受体(Melanocortin 1 receptor,MC1R,NC_030825.1:16104986-16105935)与微管蛋白3类(Tubulin-β-III,TUBB3,NC_030825.1:16104986-16116602)共用TUBB3第一外显子。TUBB3第一内含子中含有终止密码子(TGA),如在翻译过程中保留第一内含子,则翻译过程就会在第一内含子终止密码子处停止,产生一个新蛋白即MC1R所编码蛋白。
通过CPC分析、CNCI分析、pfam蛋白结构域分析和CPAT分析4种方法,共鉴定出lncRNA 10 927个(图3(c))。根据基因组上的位置关系,可将lncRNA分为基因间lncRNA(Intergenic lncRNA,lincRNA)2 585条占总lncRNA的24.4%、内含子lncRNA(Intronic lncRNA)5 356条占总lncRNA的50.6%、正义lncRNA(Sense lncRNA)2 125条占总lncRNA的20.1%和反义lncRNA(Antisense lncRNA)528条占总lncRNA的5.0%(图3(d))。基于位置关系及互补序列共预测出lncRNA 8 094条,其中基于位置关系预测出lncRNA 5 889条占比72.8%。通过与参考基因组注释的lncRNA对比分析,共鉴定出8 251个新lncRNA,这将为绒山羊基因组数据做重要补充。

(a)新转录本NR同源物种分布图;(b)新基因NR同源物种分布图;(c)转录本GO功能注释图,其中蓝色代表细胞成分,红色代表分子功能,绿色代表生物过程。(a)与(b)图例中数值表值表示该物种与NR数据库的比对注释数量,百分比为该物种比对数在总比对数中的占比。(a) Distribution map of new transcript NR homologous species;(b) New gene NR homologous species distribution map;(c) Annotated map of transcript GO function,in which blue represents cellular components,red represents molecular function,and green represents biological process.The values in (a) and (b) indicate the number of matches of the species with the NR database,and the percentage is the proportion of the number of matches of the species in the total number of matches.图2 绒山羊皮肤新转录本及基因功能注释图Fig.2 Annotation map of new transcripts and gene functions in the skin of cashmere goats

(a)基因可变剪接类型(a:Exon skipping,外显子跳跃;b:Alternative 3′ splice site,可变转录终止位点;c:Mutually exclusive exon,可变外显子;d:Alternative 5′ splice site,可变转录起始位点;e:Intron retention,内含子保留);(b)可变剪接事件数量统计图;(c)预测得到的非编码RNA韦恩图;(d)lncRNA分类图。(a) Gene alternative splicing type (a:Exon skipping,exon skipping;b:Alternative 3′ splice site,variable transcription termination site;c:Mutually exclusive exon,variable exon;d:Alternative 5′ splice site,alternative transcription initiation site;e:Intron retention,intron retention);(b) Statistics of the number of alternative splicing events;(c) Venn diagram of predicted non-coding RNAs;(d) lncRNA Sequence source pie chart.图3 绒山羊皮肤可变剪接及lncRNA统计图Fig.3 Alternative splicing and lncRNA statistics in the skin of cashmere goats
2.5 绒山羊皮肤可变多聚腺苷酸化(APA)及SSR预测
通过分析发现,SMRT测序鉴定的基因中9 485个基因至少存在1个polyA位点,151个基因存在至少5个polyA位点,平均每个基因含有1.68个 polyA位点(图4(a))。利用MEME对所有转录本polyA位点上下游50 bp的序列进行分析,鉴定得到的上下游碱基分布如图4(b)所示,绒山羊皮肤polyA剪切位点上游富含腺嘌呤,且存在保守元件AATAAA。
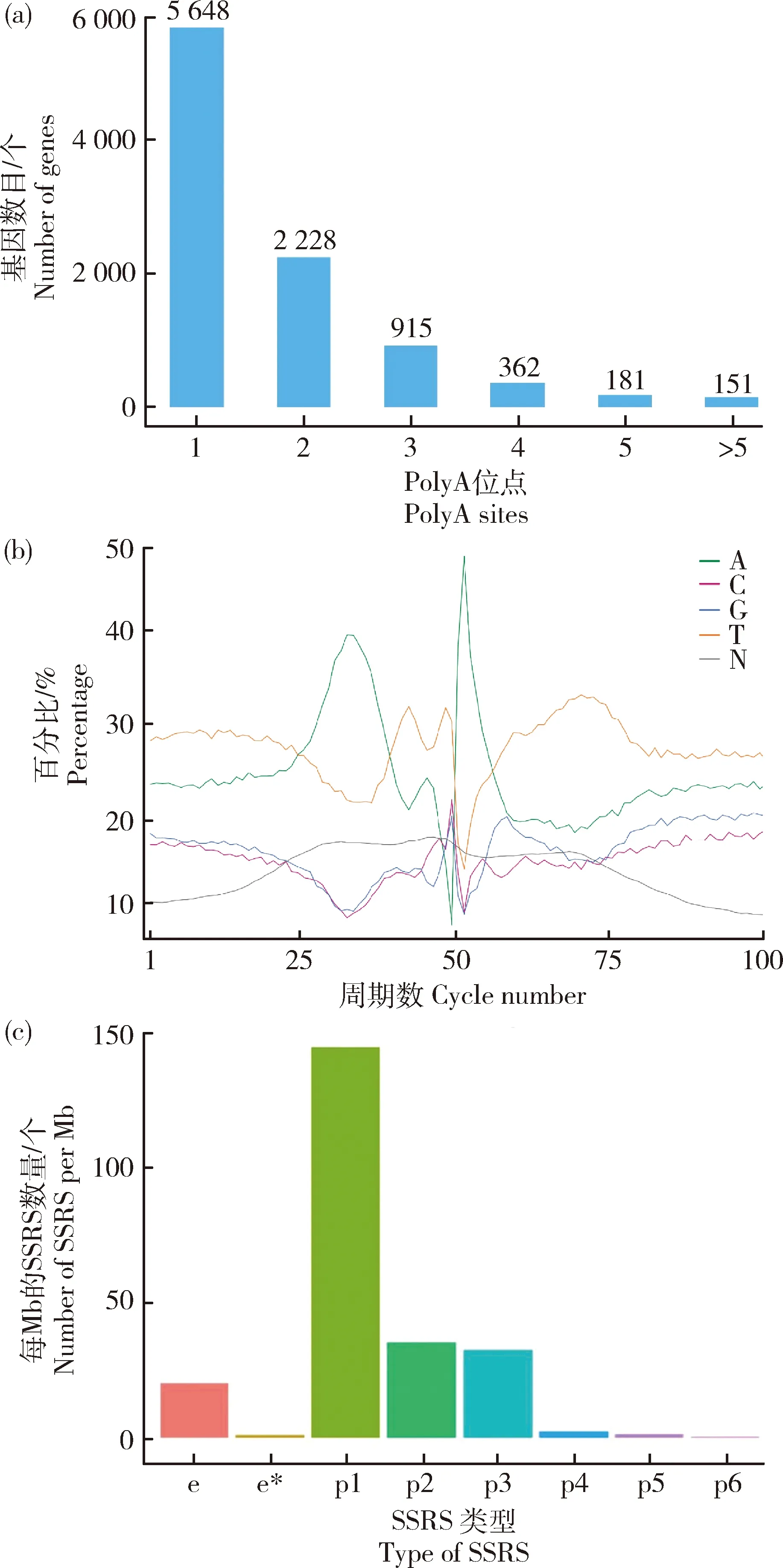
(a)ployA位点个数统计图;(b)APA上下游碱基分布图;(c)SSR类型分布图,其中包括单碱基重复(p1)、双碱基重复(p2)、三碱基重复(p3)、四碱基重复(p4)、五碱基重复(p5)、六碱基重复(p6)和混合SSR(c)。(a) Statistics of the number of ployA sites;(b) Distribution of upstream and downstream bases of APA;(c) SSR type distribution map,including single-base repeats (p1),double-base repeats (p2),three-base repeats (p3),four-base repeats (p4),five-base repeats (p5),six-base repeats (p6),and mixed SSRs (c).图4 绒山羊皮肤APA及SSR分析统计图Fig.4 APA and SSR analysis statistical chart in the skin of cashmere goats
从新转录本中筛选500 bp以上的转录本,利用MISA软件做SSR分析。共评估序列78 601条,评估序列的总碱基数为253 230 057 bp,识别的SSR总数为66 951条。鉴定出7种类型SSR:Mono-nucleotide(单碱基)42 864条、Di-nucleotide(双碱基)13 696条、Tri-nucleotide(三碱基)9 300条、Tetra-nucleotide(四碱基)706条、Penta-nucleotide(五碱基)327条、Hexa-nucleotide(六碱基)58条、Compound SSR(混合微卫星,2个SSR距离小于100 bp)6 898条,对不同SSR类型的密度分布进行统计如图4(c)。
2.6 绒山羊皮肤TF预测分析
转录因子(Transcription factor,TF)是指能够结合在某基因上游特异核苷酸序列上的蛋白质,本研究分析TF使用动物转录因子数据库animalTFDB 2.0[16],共预测得到转录因子5 641个,对不同类型的转录因子个数进行统计如图5。预测的TF类型主要是Zf-c2h2,预测数量是1 920,占总预测量的34.1%,其次是Zbtb、Homeobox以及Bhlh等,c2h2家族是已知TF家族中比较大的一类,能结合多种蛋白质发挥其功能。
3 讨 论

图5 绒山羊皮肤转录因子类型预测Fig.5 Transcription factor type prediction in the skin of cashmere goats
近年来,随着国内外绒山羊遗传育种研究的推进,绒山羊在产绒、繁殖、生长等多个重要经济性状上得到不断地改进和提升[17],然而选育绒山羊新品种、提高绒山羊优良产绒性状以及环境与基因互作仍然是现阶段研究热点。随着测序技术的进步,SMRT为分析转录组特征及mRNA 结构等工作提供了新的方法,虽然第二代测序技术比Sanger测序提供了巨大的改进,但它们的局限性不适用于某些特定的生物学问题,由PacBio开发的SMRT测序提供了一种替代方法来克服这些限制[18],与第二代测序技术不同,PacBio测序是一种实时测序方法,在读取步骤之间不需要暂停[19]。全长转录组序列对于品种选育、性状提升、基因组注释和功能研究具有非常重要的作用。虽然现阶段山羊以具有良好的遗传和基因组资源,包括高度连续的参考基因组(ARS1)[20]。然而,Muriuki等[21]认为与其他反刍动物相比,山羊参考基因组基因表达信息是有限的,因此,Muriuki等[21]构建了山羊基因表达图谱,提供了一组功能信息来注释当前参考基因组(ARS1)中15 %未注释的基因。本研究通过 SMRT 对绒山羊皮肤进行全长转录组测序分析,共获得42.41 Gb clean data的数据,获得CCS 591 585条,平均长度2 358 bp,其中FLNC序列466 058条占CCS序列的78.78%。SMRT测序技术对发现新基因与新转录本具有绝对的优势,通过SMRT测序在毛竹中发现了8 091个新转录本[22],在穿山甲中鉴定出8 014个新基因[23]。本研究测序共检测到基因18 919个,其中新基因6 166个,新发现转录本55 875个。对新发现的转录本进行序列结构分析,共预测出66 951 个SSR及10 927个lncRNA,并完成了49 573 个新转录本和4 237个新基因的功能注释。本研究所预测到的转录本数量远高于参考基因组,该数据集补充了山羊现有的遗传和基因组资源。具有高质量功能注释的高度连续参考基因组对于完善基因信息、开发畜禽物种新资源及对品种优良性状的挖掘是非常必要的。
SSR作为一种共显性遗传标记,具有特异性强、重复性好和多态性高等优点,被广泛用于遗传多样性等相关研究[24]。目前,转录组数据信息在牛科动物[25]、山羊[26]等物种中成功地开发了SSR标记。本研究中共评估序列78 601条,识别的SSR总数为66 951条,其中单碱基SSR 42 864条、双碱基SSR 13 696条、三碱基SSR 9 300条、四碱基SSR 706条、五碱基SSR 327条、六碱基SSR 58条、混合微卫星6 898条。上述对SSR分析,为进一步做绒山羊特异SSR标记及遗传图谱构建分析等研究打下了良好基础。
SMRT 测序技术还能更好的分析AS,使用该技术在绒山羊皮肤中共鉴定了25 717个AS,其中内含子保留9 899个为主要的剪接类型。Xu等[27]对山羊中的AS分析的数据结果显示,总共有14 521 个基因经历了AS,内含子保留最为普遍占AS事件总数的37.04%,与本研究AS预测结果相一致。本研究发现ClueGO将可变剪接基因富集到内皮细胞发育、mRNA分解代谢、mRNA代谢等生物学过程,同一个基因转录而来的前体mRNA通过可变剪切可形成不同的剪接异构体,最终形成不同的蛋白质而发挥不同的功能[28],提示在动物体生长发育过程中参与上述过程的基因容易发生可变剪接。结合功能富集结果及可变剪接发生位点信息发现了MC1R与TUBB3基因间存在可变剪接位点,共用TUBB3第一外显子。Herraiz等[29]研究结果表明,UV会导致人体内MC1R和TUBB3基因向MC1R-TUBB3嵌合体发生转化。当暴露于 UV 后,MC1R-TUBB3嵌合异构体会阻止cAMP途径的过度刺激[29],cAMP途径的过度刺激会导致生物体皮肤变黑,在人体内过度刺激该途径可能会增加黑色素瘤患病风险[30]。MC1R与TUBB3基因间发生可变剪接可能是山羊及绵羊在日常接触UV辐射而触发的一种自我保护机制,阻止 cAMP 途径的过度刺激减少了真黑素的产生使背负毛发颜色没有发生改变的同时又防止了UV对其产生的危害。同时,lncRNA 作为目前研究的热点,在绒山羊毛囊生长发育中起着重要的调控作用[31]。Wang等[32]基于高通量测序和生物信息学分析,在绒山羊皮肤的生长期和休止期鉴定了1 108个 lncRNA。Ge等[33]通过lncRNA观点综合分析了褪黑激素促进绒山羊次级毛囊生长。上述研究表明了lncRNA在绒山羊毛囊发育及皮肤稳态过程中发挥着不可或缺的作用。本研究通过CPC、CNCI、pfam蛋白结构、CPAT四种分析方法,共鉴定出lncRNA 10 927条,Intronic lncRNA 5 356条为主要类型,而参考基因组(ARS1)注释lncRNA数量仅为2 676条平均长度1 434 bp[20]。本研究测序获得的lncRNA预测数据远高于参考基因组及其前人的试验发现。SMRT测序鉴定的基因中9 485个基因至少存在1个poly(A)位点,预测到转录因子5 641个主要类型是Zf-c2h2,这与在巨菌草[34]及北京鸭[35]上的试验研究结果相一致。可变剪接分析中得到的新转录本对其编码区序列及对应氨基酸序列进行预测获得ORF 49 294条,其中完整ORF 39 346条,本研究获得的数据将丰富绒山羊转录组信息,同时也为绒山羊基因组做出重要的数据补充。
4 结 论
本研究对内蒙古白绒山羊皮肤样品进行了全长转录组测序并对SSR、ORF、可变剪接、新基因及新转录本进行了系统分析。试验结果为研究绒山羊皮肤mRNA全长序列、掌握完整的基因结构信息、选育绒山羊新品种和优良性状的挖掘提供了理论依据,为绒山羊基因组资源进行数据补充及提供有价值的参考。
猜你喜欢
今日农业(2022年14期)2022-09-15
北方牧业(2021年16期)2021-11-29
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
教学考试(高考生物)(2020年6期)2020-11-23
中西医结合肝病杂志(2020年2期)2020-10-27
新农业(2020年5期)2020-06-01
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
