
诺氟沙星胶囊溶出度的分析方法评价
2022-11-09 11:30于泽源刘媛媛刘小芬
广州化工 2022年19期
梁 宣,于泽源,刘媛媛,吴 琪,刘小芬
(绵阳市食品药品检验所,四川 绵阳 621000)
诺氟沙星胶囊亦称氟哌酸,是第三代喹诺酮类全合成抗菌药,为第一个氟喹诺酮类药。具有抗菌作用强,抗菌谱广,不良反应发生率低等特性,同时耐药性还未大量产生等优点[1-3],主要用于治疗敏感菌所引起的尿道、肠道等感染性疾病。现目前国内诺氟沙星单方制剂一共有802个批准文号,其中诺氟沙星胶囊有697个批准文号(规格均为0.1 g)[4]。诺氟沙星胶囊溶出的方法在国内现行的法定标准为中国药典2020年版二部[5-6],而国外英国药典(BP)[7]、美国药典(USP)[8]、日本橙皮书(JP)[9]均没有收载诺氟沙星胶囊,但收载有诺氟沙星片。胶囊剂要在体内发挥作用,前提是有良好的溶解性[10]。药物的溶解性通常用溶出度来表达,溶出度也称溶出速率,是指在规定的溶剂和条件下,药物以不同的剂型(片剂、胶囊剂、颗粒剂等固体制剂)溶出的速度和程度。溶出度是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验方法,同时也在一定程度上反映主药的晶型、粒度、处方组成、辅料品种和性质、生产工艺等的不同,造成产品质量存在差异,进一步影响临床疗效,因此溶出度是药品质量控制必检项目之一。国内现行2020年版中国药典诺氟沙星胶囊的溶出度方法与诺氟沙星片溶出度方法一致,采用紫外分光光度法测量溶出液,但近年来国内有关药品质量公告及文献显示采用现行溶出度方法发现诺氟沙星胶囊溶出度不合格的情况较多[11],不同厂家因为生产工艺等区别,法定标准检测方法结果差异较大[12]。因此针对诺氟沙星胶囊的溶出度的检测方法研究具有实际意义。
我国法定标准中诺氟沙星胶囊溶出度的测量方法是紫外可见分光光度法,而本文使用该药品法定标准中含量测定项下的高效液相色谱法与法定标准测量方法同时测定不同溶出介质中的溶出度。讨论两种分析方法的结果,尤其是处于不合格边缘值的样品,两种测试方法的结果比较分析讨论显得尤为重要。根据影响因素如溶出介质pH值、溶出反应时间t、溶出介质温度T对诺氟沙星胶囊溶出曲线进行评价,为更准确的测量结果提出建议。
1 实 验
1.1 主要试剂与仪器
RCZ-8M溶出试验仪,天津市天大天发科技有限公司;RZQ-8C溶出仪自动取样器,天津市天大天发科技有限公司;TU-1901紫外-可见分光光度计,北京普析通用仪器有限责任公司;XS205电子天平,梅特勒托利多公司;BSA224S-CW电子天平,赛多利斯科学仪器(北京)有限公司;S40H/电导率/离子综合测试仪,梅特勒-托利多仪器(上海)有限公司;Waters2695高效液相色谱仪,waters公司。
诺氟沙星对照,批号为:130450-201907,含量99.6%(质量分数为99.6%)中国食品药品检定研究院;乙腈(色谱纯),赛默飞世尔科技有限公司;冰醋酸、氢氧化钠、盐酸、磷酸、三乙胺(均为分析纯),市售;实验室用水为纯化水;诺氟沙星胶囊为同厂家,某医药集团有限责任公司,同批次(批号:210203)生产的市售品。
1.2 实验方法
溶出液的制备:是取诺氟沙星胶囊6粒,照溶出度测定法《中国药典》2020年版四部0931溶出度第二法[6],根据不同实验条件下的溶出介质,依法操作,取溶液10 mL,滤过,即得诺氟沙星胶囊溶出液。对照品溶液是取诺氟沙星对照品加对应溶出介质溶解,稀释制成约每毫升含5 g的溶液。
测定方法:紫外分光光度法参考《中国药典》2020年版测定条件为在277 nm的波长处分别测定溶出液的吸光度值。高效液相色谱法参考《中国药典》2020年版诺氟沙星胶囊胶含量测定项下色谱条件,以0.025 mol/L磷酸溶液(用三乙胺调节pH值至3.0±0.1)-乙腈(87:13)为流动相,检测波长为278 nm,测量溶出液吸光度值。
溶出曲线:溶出曲线中的溶解度值为同一影响因素下的6粒样品溶出的平均值。
2 结果与讨论
2.1 溶出介质 pH值对溶出度的影响
同批诺氟沙星胶囊在不同pH溶出介质中的溶出结果见图1。考察pH值范围从3.25~5.00之间,溶出度的变化。随着pH值的增加,溶出度值逐渐降低,当pH值在3.25~3.50时,溶出值达到短暂平衡后继续下降,说明诺氟沙星胶囊在此酸性条件下已经达到最高的溶出状态,溶出液浓度为90%以上,pH值再继续变小已没有实际意义。当在酸性条件pH4.25~4.50时,溶出度值也达到稳定的状态,pH再继续增加时,溶出度值也下降,当pH值大于5.00时,溶出度小于50%。两种检测方法显示,溶出曲线趋势一致,紫外分光光度法(UV)较高效液相色谱法(HPLC)溶出值更高,可能是HPLC具有分离作用,专属性更强,干扰更少。
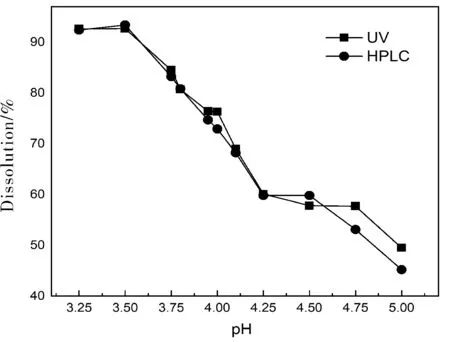
图1 不同pH值条件下紫外和液相测得的结果Fig.1 Comparison of different pH by UV and HPLC measured
通过实验显示设定诺氟沙星胶囊溶出介质的pH值不能太小,pH值太小药物很快溶解即没有生理意义,pH值太大药物溶出慢溶出效率低,也不能正确判断胶囊质量。通过紫外分光光度法和高效液相色谱法测定的结果比较,当pH值在4.0~4.1时,紫外分光光度法测定的溶出度值高于高效液相色谱法测定的结果,且紫外分光光度法测量值存在短暂平台,说明药物辅料及其它因素对紫外分光光度法的干扰更大,而高效液相色谱法中色谱柱的分离,使得测量结果受干扰情况较少。另外pH值对胶囊壳的溶解性影响较大,部分胶囊壳在pH为4.1时,已经有未溶解完全的现象,未溶解完全的胶囊势必会影响有效药物的溶解。
2.2 溶出介质的温度对溶出度的影响
同批诺氟沙星胶囊在不同温度溶出介质中的溶出结果见图2。取同一批次样品的诺氟沙星胶囊,分别置于不同温度溶出介质中,取续滤液,按照1.2项下方法,分别考察25、30、33、35、37、40、45、47 ℃的溶出值,结果见图2。当溶解介质温度在25~40 ℃时,溶解度随着温度的升高溶解度在持续增大,不同的测试方法相比,测量结果值总体差异不大;但当温度在35~40 ℃时,紫外法测量的值大于高效液相色谱法测量的值,当37 ℃时,两方法测量结果值差异最大。当温度继续增加至47 ℃时,两种方法测量的值又几趋于一致。
同时观察药物溶解状况,当温度在25 ℃时,药物还处于完好状态,只是将胶囊壳软化,从溶出度结果来看,药物几乎未溶解在介质中。随着温度的升高,溶出度越来越大。溶出度的温度是模拟人体温度来设置的,人体正常温度为36.5~37.3 ℃,因此溶出温度为(37±0.5)℃,太高或太低都无生理意义。对比两种方法的测量比较,温度升高有利于药物溶出,溶解过程属于吸热过程。
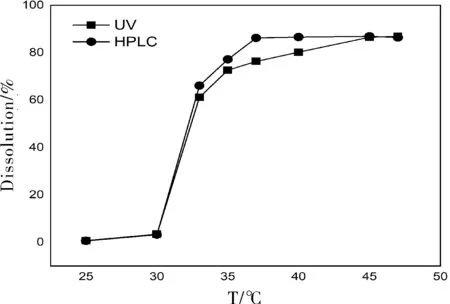
图2 不同温度下用紫外和液相条件下测得的溶出度Fig.2 Comparison of different temperature by UV and HPLC measured
2.3 不同溶出反应时间条件下的药物溶出度
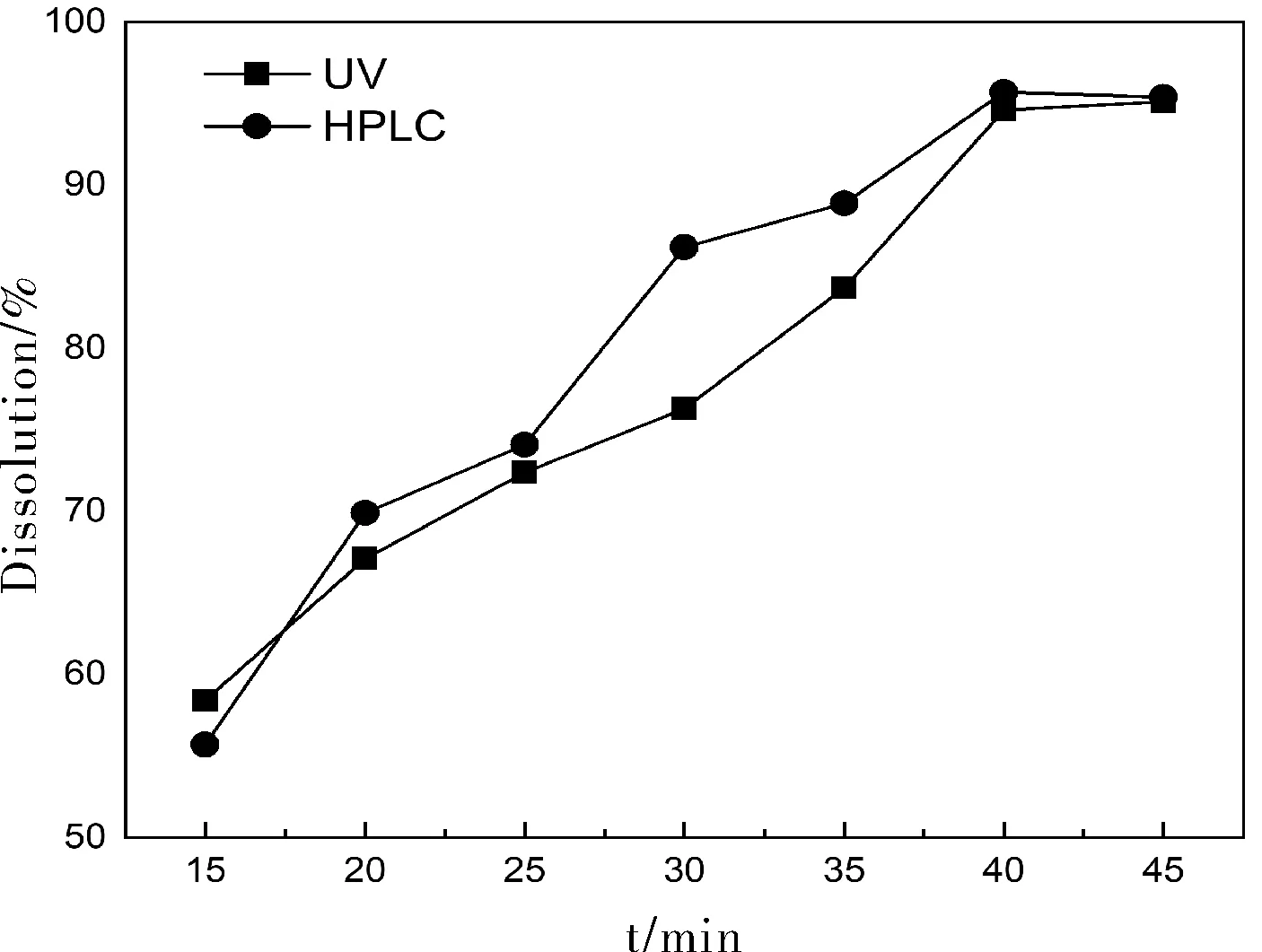
图3 不同时间紫外和液相测定的溶出度Fig.3 Comparison of different time by UV and HPLC measured
取同一批次样品的诺氟沙星胶囊,分别置于溶出介质中,控制溶出反应时间,分别在15、20、25、30、35、40、45 min时,取续滤液,按照2.1项下方法,测量并绘制溶出曲线,结果见图3。当溶出时间至15 min时,已测得溶出度为55%左右,当时间到30 min时两种测量方式的结果数据差距最大,整个时间段内,高效液相法测定值明显高于紫外分光光度法测定值,至40 min时,溶解已趋于平衡,且两种方式的测量值差异较小。
3 结 论
通过以上对诺氟沙星胶囊溶出曲线的研究发现,紫外分光光度法和高效液相色谱法两种方法测得溶出曲线变化趋势一致,但数据结果有差异。紫外分光光度法较高效液相色谱法易受到某些杂质、辅料、胶囊壳的溶解性等因素影响。液相色谱分析法中有分离的步骤,所以在相同的溶出反应条件下液相色谱法相比紫外分光光度法测量更准确。溶出度受溶出介质pH值和温度的影响很大,当pH值为4.0附近时,溶出度值差距较大。因此在配置介质溶液时,一定要确保溶液pH准确。当溶出介质pH值为4.0时,不同的反应时间和不同的反应温度都显示出液相色谱分析法的测得值更高。当不合格样品的溶出度值处于不合格边缘值时,在排除其它因素的影响后,应考虑使用高效液相色谱法对溶出液进行再次测量,综合判断后给出判定。
综上,诺氟沙星胶囊溶出度的测定中,pH值为4.0、温度为37 ℃、反应时间为30 min时,若出现溶出度临界值影响结果判断时,应利用该品种含量测定项下的高效液相色谱法测定续滤液,综合判定,其结果值能更为客观的反应该药物溶出情况。
猜你喜欢
中国畜牧业(2022年21期)2022-12-01
昆钢科技(2022年2期)2022-07-08
口腔护理用品工业(2021年4期)2021-11-02
昆明医科大学学报(2021年8期)2021-08-13
广东蚕业(2020年6期)2020-09-24
智富时代(2018年6期)2018-08-06
智富时代(2018年6期)2018-08-06
神州·下旬刊(2017年6期)2017-10-28
药学研究(2015年11期)2015-12-19
中国高新技术企业(2015年24期)2015-06-25
