
低射频功率-电感耦合等离子体质谱法测定高纯石英样品中痕量钾
2022-11-13 05:06张海啟倪文山刘磊张宏丽
矿产保护与利用 2022年4期
张海啟, 倪文山,刘磊,张宏丽
1.中国地质科学院郑州矿产综合利用研究所,河南 郑州 450006;2.国家非金属矿资源综合利用工程技术研究中心,河南 郑州 450006
引 言
高纯石英作为世界稀缺、我国短缺的高技术战略矿物资源,是半导体芯片、太阳能光伏、光纤通信、航空航天、精密光学和特种玻璃等高新技术产业不可缺少且无法替代的重要功能性材料,是真正“卡脖子”的矿物资源[1-4]。
石英化学成分较为单一,主要成分是SiO2,但大多数石英样品中含有铝、钛、钠、钾、锂、钙、镁、铁、铬、镍、铜等杂质元素,同时还有一定量的-OH存在。石英化学成分变化中最重要的机制就是铝以类质同象的形式取代硅,而Al3+取代Si4+所造成的电荷不平衡,由半径较小的碱金属元素离子Li+、Na+、K+进入间隙的位置进行电荷补偿[5]。因碱金属元素在高温下起着助熔剂的作用,使高温玻璃出现失透、高温变形等不良现象,影响石英制品的热稳定性、光学特性、热学特性等。因此,我国已经制定了20余项石英行业现行国家标准、行业标准和地方标准,对不同产业用石英砂的粒径、透明度、二氧化硅含量、烧失量、杂质元素含量分别进行了规定[6-11]。《GB/T 32649—2016 光伏用高纯石英砂》规定杂质元素总含量应小于或等于25 μg/g,其中钾、锂、钠含量总和小于2.5 μg/g,单项含量不超过1.0 μg/g。
钾元素作为石英砂产品中严格限定的杂质元素,目前采用的分析方法主要有电感耦合等离子体发射光谱法(ICP-AES)[12]和原子吸收光谱法(AAS)[13]。电感耦合等离子体质谱法(ICP-MS)具备精确测定纳克/ 升(ng/L)甚至更低浓度元素含量的能力,是最适合测定痕量及超痕量无机元素的分析测试设备[14-16]。而钾元素在质谱测定中因质量数较小存在质谱干扰因素多的问题, 常规的标准测定模式无法实现痕量钾元素的准确测定。目前,GB/T 32650—2016“电感耦合等离子体质谱法检测石英砂中痕量元素”阐述了采用ICP-MS测定了高纯石英样品中钾元素[9]。但该国标方法仅给出基本操作流程,未对具体元素测定的干扰消除方式进行说明,在日常生产测定中仍需根据自身测定要求,进行不同质谱测定条件的选择和优化。常规的测定条件下,氩、氧、氢离子会与样品溶液中的酸基体相结合,对待测元素产生多原子离子干扰[16]。基于此,本文尝试通过降低质谱测定中射频功率,即采用冷等离子体,有效避免质谱中多原子离子的质子化,从而避免钾元素在测定中受到多原子离子的干扰,以获得较低的检出限和更高的稳定性。
1 试验部分
1.1 主要仪器及工作条件
ICAP Q型电感耦合等离子体质谱仪(Thermo Scientific);质谱仪的工作条件见表1。
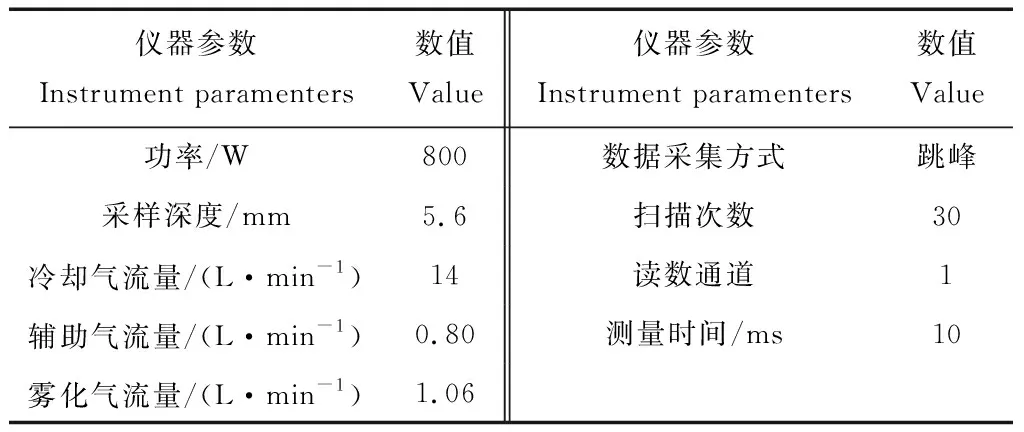
表1 ICP-MS仪器工作参数
1.2 主要试剂
钾元素标准储备溶液(1.000 0 mg/mL)、GNM-M 170218—2013多元素标准溶液(Al、B、Ba、Be、Ca、Cr、Cu、Fe、K、Li、Mg、Mn、Na、Nb、P、Ta、Ti,100 μg/mL)、GNM-M 080129—2013多元素标准溶液(B、Ga、Li、Pb、Sb、Sr、Tl、Zr)购置于国家有色金属及电子材料分析测试中心;Rb标准储备溶液(500 μg/mL):准确称取0.078 1 g Rb2SO4高纯试剂于100 mL玻璃烧杯中,加入10 mL水溶解样品后,完全转移至100 mL容量瓶中,定容、摇匀;采用逐级稀释的方法配得Rb内标溶液(50 ng/mL)。
Tune B iCAP Q 溶液(Thermo Scientific):Ba、Bi、Ce、Co、In、Li、U各元素的质量浓度均为1.000 0 μg/L,介质为2%硝酸(体积分数)-0.5%盐酸(体积分数),购置于赛默飞世尔科技(中国)有限公司;iCAP Q/Qnova 校正溶液(Thermo Scientific),购置于赛默飞世尔科技(中国)有限公司。
试验所用硝酸、盐酸、氢氟酸均为电子级酸试剂,购置于天津市科密欧化学试剂有限公司;试验用水为产自Milli-Q系统(默克密理博,法国)的超纯水(电阻率=18 MΩ·cm,25 ℃)。
1.3 试验方法
高纯石英样品须经105~110 ℃干燥,而后准确称取1.000 0~2.000 0 g样品于30 mL具盖聚四氟乙烯坩埚中,少量超纯水润湿样品后,加入8 mL 氢氟酸、1 mL硝酸,加盖、置于110 ℃电热板上浸取2 h;取下稍冷后去除坩埚盖,将坩埚置于180 ℃电热板上加热冒烟,除尽酸试剂;待白烟冒尽后,将坩埚取下冷却,加入1 mL硝酸、2 mL超纯水,加盖置于电热板上微沸取下;将样品溶液完全转移入100 mL聚全氟乙丙烯(FEP)容量瓶中,定容、摇匀;在选定的仪器条件下,由三通在线加入100.0 ng/mL的铷内标,采用ICP-MS测定溶液中钾元素的含量。
2 结果与讨论
2.1 称样量优化
高纯石英砂采用高品位石英矿经选矿、煅烧、水淬等工艺而得,最终高纯石英产品中几乎不存在矿物包裹体,而杂质元素基本赋存于晶格中或者气液包裹体中,呈现分别不均匀分布的特征。因此,为保证测试过程中取样的代表性,高纯石英样品的称样量相较常规岩石矿物分析要有所增加,同时大的取样量也能够提高样品溶液中待测元素含量,进而提高测试设备的响应信号。但同时也需考虑不同仪器设备对基体浓度的耐受性。因此,样品的称样量直接影响待测元素测定的准确性。基于此,本文试验了0.100 0 g、0.500 0 g、1.000 0 g、1.500 0 g、2.000 0 g、2.500 0 g、3.000 0 g不同称样量条件下典型高纯石英样品内钾元素测定结果的稳定性,试验结果如图1所示。由称样量优化结果可知,随着称样量的增大,测定结果稳定性增强。但当称样量大于2.000 0 g时,由于测定设备对基体耐受性有限,需对样品溶液进行稀释、分取,从而导致测定结果波动性变大。同时,参考现行高纯石英相关国家标准分析方法、行业标准分析方法中取样量的要求,称样量最终优化结果为1.000 0~2.000 0 g之间。
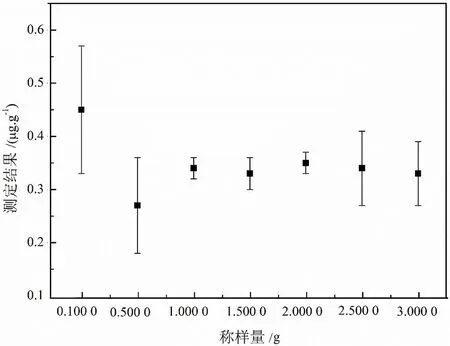
图1 高纯石英中钾杂质测定的称样量优化
2.2 酸试剂用量优化
项目组前期建立了高纯石英中杂质元素铁的测定方法[15],选用氢氟酸+硝酸(10+1)mL为溶解试剂、硝酸为提取试剂的试剂组合方式进行了样品溶解。基于当下“双碳”战略倡导绿色、环保、低碳的生产生活方式,本文在前期研究基础上,进一步探索了更小的酸试剂用量下样品的溶解情况。在1.000 0 g称样量前提下,分别加入不同体积溶解试剂和提取试剂,测定不同试剂体积下样品的溶解性及钾元素的测定结果稳定性,结果如表2所示。由表中测定数据对比可知,将溶样过程中氢氟酸用量降低至7 mL 及以下时,在除酸过程中因试剂量小、酸试剂除尽后坩埚底部有少许未溶样品颗粒,溶样不完全;当氢氟酸试剂用量大于8 mL时,样品溶解完全且测定结果稳定。因此,最终选择氢氟酸+硝酸(8+1) mL作为样品化学前处理的试剂用量。

表2 样品化学前处理中酸试剂用量优化
2.3 同位素选择
钾元素有39K、40K和41K三个同位素,其中39K和41K两种为稳定的同位素,丰度分别为93.26%和6.73%;40K为放射性核素,丰度为0.01%,各同位素的丰度及可能存在的质谱干扰情况见表3。由表中结果可知,由于Ar-H离子的形成,39K和41K两种钾元素稳定的同位素测定均会受到明显的多原子离子产生的质谱干扰。特别是在质谱测定中的常规射频功率1 550 W高于所有金属离子的第一电离能,导致该功率下多原子离子以及双电荷离子产率较高,造成常规射频功率下质谱仪难以实现钾元素的准确确定。因此,本文选择丰度高的39K稳定同位素作为钾元素的测定同位素,而后探索抑制多原子离子干扰的方法,以实现待测元素的准确测定。
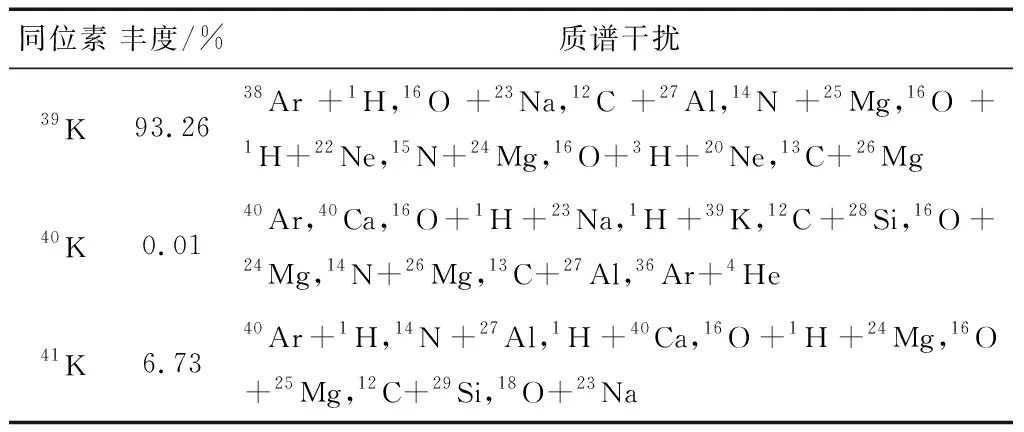
表3 钾元素同位素丰度及可能存在的质谱干扰
2.4 射频功率优化
为有效降低质谱测定钾元素时存在的多原子离子干扰,本文采用降低质谱测定中的射频功率(即采用冷等离子体的方式),根据钾元素的第一电离能(4.34 eV)远低于Ar(15.76 eV)这一特点,保证钾元素离子化的同时尽量减少38Ar1H+的生成,以最大程度减少对39K同位素测定的干扰,实现待测溶液内超痕量钾元素的准确测定。
ICAP-RQ型质谱仪提供的冷焰模式等离子体焰的温度低,电离电位较高的Ar的电离度明显降低。本文采用电阻率为18 MΩ·cm(25 ℃)的超纯水进行质谱测定中荷质比(m/z)为39背景强度研究,因超纯水内几乎不含导电离子,因此该荷质比下的质谱信号强度即代表了测定中38Ar1H+干扰的背景强度。
试验研究结果如图2所示,随射频功率降低,超纯水中测定的m/z=39处信号强质量数处质谱峰强度显著降低;当射频功率低于840 W时,质谱信号强度变化趋势变缓,射频功率在840~800 W之间,质谱信号强度差异较小;该结果表明当射频功率在840 W以上时,质谱背景中38Ar1H+背景峰仍较明显;当射频功率降低到840 W以下, 质谱背景中38Ar1H+的离子强度明显降低,当射频功率在800 W时,m/z=39处的质谱信号强度值约为射频功率1 550 W时质谱强度的0.000 1%,由此可见钾元素测定中背景产生的质谱干扰得到了有效抑制。而钾元素作为第一电离能最低的元素,在低功率条件下该元素的电离度仍能够满足检出需求。因此,后续测定均采用800 W射频功率进行测定。

图2 不同射频功率下m/z=39信号强度变化图
2.5 内标元素选择
内标法利用待测元素与内标元素在分析仪器上对应的信号比来补偿分析信号的波动以提高测定的精密度和准确度。质谱测定中内标元素的选择标准为:(1)质量数与被测元素接近;(2)第一电离能与被测元素接近。基于这两点,同时充分考虑本文测试的石英样品组分特征,最终选择85Rb元素作为内标元素,来校正质谱测定中基体效应的影响。
2.6 质谱采样深度优化
采样深度表示质谱设备中采样锥孔与焰炬的距离,是影响质谱测定灵敏度的一个重要参数。ICAP RQ标准模式下设备采用的采样深度为5.0 mm,本文因采用冷焰模式,降低了射频功率,一定程度上降低了待测元素的灵敏度。因此本文对冷焰模式下的采样深度进行了优化。以10 ng/mL的钾元素标准溶液为待测液,进行不同采样深度条件下待测元素测定结果的稳定性试验,结果表明,当采样深度调整为5.6 mm时,待测元素的测定结果稳定性最佳。因此,最终本文采用的采样深度为5.6 mm。

表4 质谱采样深度优化
2.7 雾化气流速优化
雾化气流速大小直接影响雾化器提升量、雾化效率、雾滴粒径、气溶胶的停留时间,进而对测定结果造成直接影响。因此,本文对试验中的雾化气流速进行了优化,结果如图3所示。图示结果表明,适当提高雾化气流速能够提高待测元素的测量灵敏度,但过大的流速反而降低测定结果的稳定性。本文最终优化后的雾化气流速为1.06 L/min。
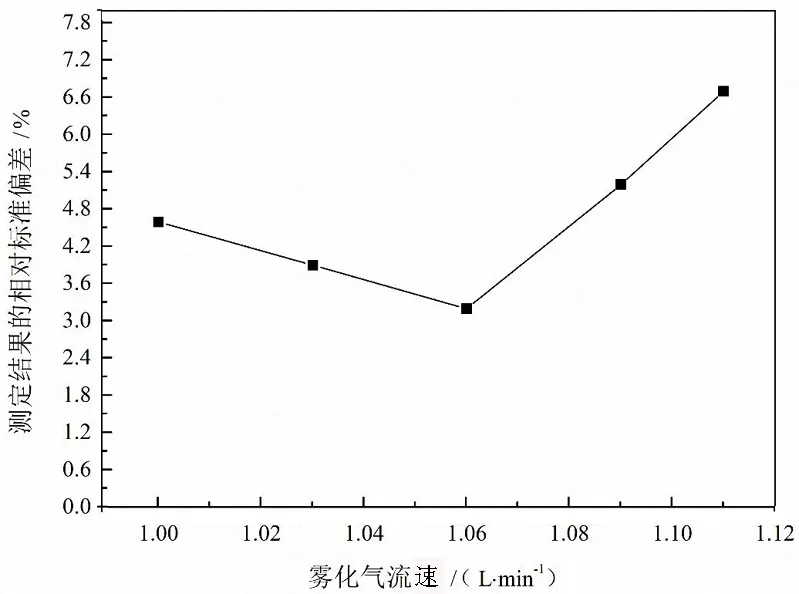
图3 雾化气流速优化
2.8 校准曲线和检出限
在选定的试验条件和仪器测定条件下,在线加入50.0 ng/mL85Rb为内标,测定钾元素标准系列溶液。以钾元素质量浓度为横坐标,质谱强度为纵坐标绘制校准曲线。结果表明,钾元素质量浓度在0.100~50 ng/mL范围内与其质谱强度呈线性相关,线性回归方程为I(cps)=1009ρ(ng/mL)+6257,相关系数r=0.999 6。平行测定11份空白溶液,以1.000 0 g称样量计,以3倍空白标准偏差计算方法的检出限为0.057 μg/g,10倍空白标准偏差计算方法的定量测定下限为0.191 μg/g。
2.9 样品分析
因我国目前尚无高纯石英(SiO2含量在99.99%以上)相关国家标准物质,为验证试验方法的准确性和可靠性,本文选取了7个不同钾含量水平的高纯石英产品(其中样品1~2为进口高纯石英砂产品,产品 3~4为国内高纯石英砂产品,样品5~7为本研究单位内部选冶加工后的高纯石英样品),按照上述试验方法对每个样品进行7次平行测定,并进行加标回收试验。
同时采用GB/T 3284—2015规定的试料溶液中钾元素在10-9级时的测定方法—石墨炉原子吸收光谱法(GF-AAS) 进行方法对照,结果见表4所示。由表中结果可知,测定结果的相对标准偏差(RSD)在3.1%~5.7%之间;加标回收率在98.3%~109.2%之间,满足国家地质矿产行业标准DZ/T 0130—2006的要求[17](待测元素质量分数在1.010-6~1.010-4时,加标回收率范围应在为90%~110%之间);本试验方法测定值和石墨炉原子吸收光谱法的测定结果基本一致。

表5 高纯石英样品中钾的测定结果
3 结 论
基于痕量碱金属元素钾的准确测定在高纯石英产品质量评价中的重要作用,本文通过降低质谱测定中的射频功率,即采用冷等离子方式,建立了一个基于常压混酸溶样-电感耦合等离子体质谱法测定高纯石英中痕量钾的测定方法。通过样品化学前处理试验参数和仪器测定参数的优化,在称样量1.000 0 g、射频功率800 W、采样深度5.6 mm、载气流速1.06 L/min的最优条件下,以50 ng/mL 铷为内标元素,经测定钾质量浓度在0.100~50 ng/mL范围内与其质谱强度呈线性相关,相关系数为0.999 6。以1.000 0 g称样量计,本方法对高纯石英中钾的检出限为0.057 μg/g,定量限为0.191 μg/g。选择典型商品化高纯石英样品进行本方法的准确性和精密性试验,结果满足国家地质矿产行业标准DZ/T 0130—2006的要求。同时与GF-AAS法进行了方法比对,测定结果一致。试验结果充分证明本方法适用于高纯石英样品中痕量钾的测定。本文为高纯石英产品品质的准确评价、产品开发提供了重要的方法支撑。
猜你喜欢
分析仪器(2022年5期)2022-10-14
中国石油大学学报(自然科学版)(2022年4期)2022-09-05
矿产保护与利用(2022年5期)2022-03-28
化工管理(2021年7期)2021-05-13
矿产综合利用(2020年1期)2020-07-24
山东冶金(2019年6期)2020-01-06
中国宝玉石(2019年4期)2019-09-03
电子制作(2018年14期)2018-08-21
中国继续医学教育(2015年1期)2016-01-06
铁道科学与工程学报(2015年5期)2015-12-24
