
T型钙通道在心血管疾病中的研究进展*
2022-12-03 08:46张弛赵爽陈露岚胡亦清申程
中国病理生理杂志 2022年11期
张弛,赵爽,陈露岚,胡亦清,申程
(1济宁医学院附属医院,济宁医学院,山东 济宁 272067;2上海市徐汇区中心医院体检科,上海 200031;3复旦大学附属中山医院心内科,上海市心血管病研究所,上海 200032;4济宁医学院附属医院心内科,济宁市心血管疾病诊疗重点实验室,山东 济宁 272067)
电压门控钙通道(voltage-gated calcium channels,VGCC)能够参与调节多种可兴奋性细胞和非兴奋性细胞的生理活动,其中T型钙通道在神经元中产生低阈值钙尖峰并影响动作电位放电模式,在神经递质释放、树突共振现象及基因表达调控中起着关键作用[1]。在血管平滑肌细胞中调节肌源性张力,在内分泌细胞中调节激素的分泌,以及在精子中调节顶体反应。重要的是,T型钙通道在心脏细胞中能够影响心脏的起搏和脉冲的传导[2-3]。近年来,对T型钙通道活性的分子机制和作用靶点的研究不断取得新的进展,转基因动物实验已经证明T型钙通道是治疗包括心肌肥大等多种心血管疾病的重要药物靶点,其活性受多种激素的影响。因此,关注T型钙通道在心血管疾病中的作用机制对于心血管疾病的治疗和相关药物的研发具有重要意义。
1 VGCC
VGCC激活并介导细胞外Ca2+内流进入细胞质。作为第二信使,Ca2+与许多细胞反应有关,包括神经递质释放、肌肉收缩和基因表达。因此VGCC是电兴奋的关键信号传感器,能将细胞膜上的电信号转化为细胞内具有重要生理意义的Ca2+瞬态[4]。
1.1 功能心肌细胞膜上存在2种不同的VGCC(L型和T型)。VGCC具有3种依赖电压的不同功能状态:关闭或休眠通道、激活或打开通道和失活通道。去极化时,激活通道在几毫秒内从静止状态转变为激活状态,然后迅速失活。然而,不同于Na+和K+主要通过膜电位改变的细胞膜流动交换方式,Ca2+通过VGCC进入细胞并与许多细胞反应相耦合。在心肌和平滑肌细胞中,VGCC通过增加细胞质中Ca2+浓度直接引发收缩而激活,或者通过肌浆网中对雷诺丁受体(ryanodine receptor,RyR)敏感的Ca2+释放通道间接激活钙依赖钙释放机制[5-6]。
1.2 亚型编码VGCC的α1亚基目前已分出10种。按照α1亚基的基因型,可分为Cav1、Cav2和Cav3这3个家族,其中Cav1包含Cav1.1~1.4四个亚型,Cav2包 含Cav2.1~2.3三 个 亚 型,Cav3包 含Cav3.1~3.3三个亚型[7]。Cav1亚家族启动神经元的收缩、分泌、调控基因表达,突触的转运整合和特殊感觉细胞带状突触的传递。同时,Cav1在启动心脏和平滑肌收缩中发挥重要作用,Cav1中含有一个形成孔的α1D亚基,是正常心脏起搏所必需的[8]。Cav2亚家族主要负责在快突触中启动突触传递;Cav3亚家族则对于节律放电细胞(如心肌细胞和丘脑神经元)动作电位的重复放电非常重要[9]。
1.3 分类VGCC按照电压激活特性分类,可分为高电压激活型(high-voltage activation,HVA)和低电压激活型(low-voltage activation,LVA),HVA通道在膜去极化程度大时响应激活,而LVA通道在低电压变化时即可激活,此时的膜电位与可兴奋性细胞静息膜电位相近[10]。按照Ca2+电流门控特性分类,又可分 为L型(Cav1.1~1.4)、P/Q型(Cav2.1)、N型(Cav2.2)、R型(Cav2.3)和T型(Cav3.1~3.3)[10]。
2 T型钙通道
2.1 结构和分子特性L型钙通道是由α1核心亚基和附属亚基(包括α2δ、细胞内β和跨膜γ)组成的异源四聚体。α1亚基由4组同源重复组成,每组重复包含6个跨膜片段(S1、S2、S3、S4、S5和S6)以及S5与S6之间形成的连接环。S5、S6及连接环共同形成通道的孔道,是药物调节钙通道的结合位点。每组重复中S4作为电压感受域,在膜去极化时响应使通道打开[7],辅助亚基调节α1亚基的通道动力学、门控特性以及通道上膜运输。2019年,颜宁团队发表在《Nature》上的文章首次揭示了人类T型Cav3.1通道高分辨冷冻电镜结构,提出与L型亚基不同的是,T型钙通道的α1亚基可以自主支持通道功能。此外,由于Cav3.1的α1亚基上2个门控相关残基不同于L型Cav1.1通道,只需更少的能量来激活通道胞内门控开放,促进Cav3.1在低电压下激活。通道的高分辨率结构分析有助于对其独特功能的机制研究,并通过引导通道激动剂或拮抗剂的结构研发药物[11]。
Cav3亚家族传导T型Ca2+电流。这种Ca2+电流在相对负的膜电位下被激活,与大多数细胞中的Na+电流范围相同。而且与其他Ca2+电流相比,它们具有快速的电压依赖性失活[12]。因为它们在负膜电位下被激活,而在负膜电位下Ca2+进入的驱动力很大。因此,这种Ca2+电流非常适合按一定的频率触发动作电位,它们同样也很适合产生大量的Ca2+瞬态。通过cDNA克隆和测序鉴定得出Cav3是一个由3种α1亚基组成的家族。值得注意的是,这些钙通道亚基具有与Cav1和Cav2通道相同的分子组织,但是只有25%的氨基酸序列相同,这表明钙通道的亚家族在进化时就会彼此分离[9]。
尽管Cav3通道与Cav1和Cav2通道具有相似的结构,但目前没有明确的证据表明它们与同一组的辅助亚基相互作用。事实上,普遍的观点认为α1亚基独立于其他亚基发挥作用,这在钠通道和钙通道家族中是独一无二的[9]。
2.2 组织空间表达T型钙通道开放时间较短,与低膜电位下Ca2+的跨膜运动有关,在窦房结(sinoatrial node,SAN)细胞早期除极(起搏电流)中起特殊作用。在胚胎期时,T型钙通道在整个心脏广泛表达,表明其在胎儿生命的特定阶段的重要作用,但是出生后在心室肌细胞表达迅速下降。在成人心脏中,仅在心脏传导系统中的起搏细胞可以观察到高密度的T型钙通道[13]。成人心脏SAN、房室结(atrioventricular node,AVN)和浦肯野纤维中保留Cav3.1和Cav3.2表达,提示其参与心脏自动性和传导[14],并在SAN细胞早期除极中起到起搏电流的作用。研究表明,Cav3.2缺陷小鼠窦房节律正常,而Cav3.1缺陷小鼠SAN恢复时间延长,SAN细胞起搏器活动减慢,心率减慢,房室传导延迟[15]。这些结果表明,Cav3.1是心脏心律产生的主要T型钙通道参与者[16]。此外,在血管平滑肌中分布的T型钙通道有维持血管平滑肌张力的作用[17]。
3 T型钙通道与心血管疾病
3.1 T型钙通道对心脏和血管功能的调节
3.1.1 T型钙通道参与维持心脏自律性心脏自律性是指由于相对缓慢的舒张期去极化的存在,起搏细胞在SAN中产生自动化,它驱动膜电压从复极化阶段的结束直到到达下一个动作电位的阈值。之后SAN脉冲传导到心房和AVN,并通过浦肯野纤维网传播到心室,推动整个工作心肌的收缩[18]。心脏的起搏细胞主要位于SAN,将SAN起搏器细胞表面膜上离子通道的集合设想为一个表面的“膜时钟”[19]。在舒张后期去极化过程中,肌浆网通过RyR产生释放Ca2+,这些Ca2+被称作是细胞内的“Ca2+时钟”。在自动激发的SAN细胞中,膜时钟和Ca2+时钟不是单独工作,而是通过膜电压、肌膜下Ca2+、蛋白激酶A(protein kinase A,PKA)和磷酸化的钙/钙调蛋白依赖性蛋白激酶II(calcium/calmodulin-dependent protein kinase II,CaMKII)等相互作用,两个子系统时钟相互约束,形成一个耦合的时钟模型,驱动正常心脏起搏器细胞的自动性[20]。SAN起搏器细胞同时表达T型和L型钙通道。T型钙通道的异构体Cav3.1和Cav3.2的mRNA在SAN中表达,成人SAN中主要的T型钙通道亚型是Cav3.1。Cav3.1介导的钙通道电流的激活阈值为-55 mV。然而,只有10%的稳态Cav3.1介导的钙通道电流在最大舒张电位时可用。Cav3.1介导的钙通道电流稳态激活和失活曲线的相对位置不能表明有显著的窗口电流分量[16]。此外,T型钙通道已经在SAN、AVN和浦肯野纤维这3个心脏节律中心中被观察到[21],表明T型钙通道可能与舒张期去极化的产生有关。
Chen等[15]通过靶向灭活Cav3.2和Cav3.1的转基因小鼠阐明T型钙通道异构体在心脏起搏和脉冲传导中的作用。与野生型小鼠相比,通过同源基因靶向技术构建的Cav3.2缺陷小鼠,其心率和心电图波形无显著差异,表明T型钙通道中的Cav3.2不能显著促进心脏自动性的产生和传导。与Cav3.2缺陷小鼠相反,小鼠的Cav3.1基因失活导致SAN传导速率中度降低和房室间隔传导延长。Cav3.1通道相关的通道病强调了钙通道在心脏自动性的产生和调节中的重要性[22]。这些研究说明在心脏自律性的产生和传导中,T型钙通道的Cav3.1起主要作用。
3.1.2 T型钙通道缺陷导致先天性心脏传导阻滞(congenital heart block,CHB)发生CHB是一种被动获得的自身免疫病,发生于患有风湿性疾病的母亲的妊娠期,并与母亲抗Ro/SSA和抗La/SSB抗体有关。该病的特征是房室传导阻滞,可在妊娠16~25周的胎儿中检测到[23]。Cav3.1缺陷小鼠出现SAN传导速率中度降低和房室间隔传导延长,这是CHB的一种表型[24]。这提示Cav3.1在CHB的发展过程中可能与抗Ro抗体阳性IgG发生交叉反应[25]。Strandberg等[26]观察到人类孕18~22.6周胎儿心脏AVN中T型钙通道Cav3.1的mRNA表达与心尖区存在差异。实验结果表明CHB胎儿心肌细胞表面可表达Cav3.1,并影响妊娠的孕妇血清对α1G蛋白的反应性。CHB孕妇血清对α1G的反应显著高于对照组,反应性的表位被定位到一个p305的肽段(对应于连接跨膜片段S5-S6的细胞外环aa305-319)。单细胞膜片钳电生理学实验也证实了CHB母体血清会不可逆地降低小鼠SAN细胞T型Ca2+电流。因此CHB母体血清抗体容易靶向人类胎儿心肌细胞中T型钙通道Cav3.1的外表位。
自身抗体在Cav3.1[26]和Cav1.3[27]通道α1亚基上存在结合位点。在这2个通道中,结合位点都位于连接α1亚基的S5和S6域胞外环路,靠近通道孔。Cav1.3和Cav3.1通道中结合位点的定位与观察到急性灌注自身抗体能迅速抑制Cav1.3介导的L型钙通道和Cav3.1介导的T型钙通道的结果一致。人类胎儿心脏CHB的发生可能涉及2个阶段:在第一阶段,母体自身抗体结合α1亚基抑制Cav1.3和Cav3.1通道;在第二阶段,VGCC在肌细胞中内化,同时这也是不可逆的阶段。此外,有研究也提出长期暴露于母体自身抗体和通道内化可引发肌细胞死亡而导致CHB的产生,并在患者死后的心脏中检测到SAN和AVN的纤维化和钙化[27]。
L型Cav1.3和T型Cav3.1钙通道在心脏自动性的产生中起重要作用。自21世纪以来,转基因小鼠已经帮助确定了离子通道亚型在起搏器活性中的作用[16]。重要的是,这些小鼠系再现了由于Cav1.3通道突变和Cav3.1通道自身免疫功能丧失而导致的某些形式的遗传性SAN病变。
3.1.3 不同亚型T型钙通道对心肌肥大的作用不同心脏能够通过增加肌肉的质量从而应对压力的变化,但长期处于压力超负荷下心脏会出现一种常见的适应性反应,称为心肌肥大,其持续进展会导致心力衰竭[28-29]。心肌肥大的特征包括细胞大小改变、肌动蛋白重组、蛋白质合成增加和胎儿基因重新表达。在肥大心脏中重新表达的胎儿基因包括心房钠尿肽、脑钠肽、收缩蛋白的胎儿亚型(α-骨骼肌肌动蛋白和β-肌球蛋白重链),以及胎儿心脏中表达的离子通道,如超极化激活的环核苷酸门控通道和T型钙通道[30]。长期心肌肥大可能会导致心力衰竭和死亡,病理性心肌肥大通常与心脏的结构和功能重塑有关,其中参与心脏调节的钙通道发挥重要的作用[31]。
目前不同亚型的T型钙通道在心肌肥大中的作用不同。Chiang等[32]提出,Cav3.2通过激活钙调磷酸酶(calcineurin)/活化T细胞核因子(nuclear factor of activated T cells,NFAT)途径参与心肌肥大的发病机制。在缺乏Cav3.2的小鼠中,压力超负荷诱导的肥大受到严重抑制,但在缺乏Cav3.1基因的小鼠中却没有明显表现。这一结果说明Cav3.2通过激活calcineurin-NFAT信号级联调节病理性心肌肥大,并对心肌肥大的产生起重要作用。而Nakayama等[33]却指出Cav3.1能够对抗心肌肥大的发生和发展。通过对具有可诱导表达Cav3.1的心脏特异性转基因小鼠与Cav3.1缺陷小鼠进行压力超负荷、异丙肾上腺素输注和运动诱导等,结果显示Cav3.1转基因小鼠对心肌肥大具有抵抗力,没有产生心脏的病理表现,而在Cav3.1缺陷小鼠中却表现出增强的肥大反应。在机制上,Cav3.1保护心脏是通过调节一氧化氮合酶3(nitric oxide synthase 3,NOS3)实现的,NOS3是一种钙激活的信号效应器,已知可改变心脏肥大反应。Cav3.1与NOS3相互作用,后者在压力超负荷后增加了Cav3.1转基因小鼠心脏中环鸟苷酸(cyclic guanosine monophosphate,cGMP)依赖性蛋白激酶I型的活性,从而实现Cav3.1对抗心肌肥大的作用[34]。这些实验结果表明Cav3.1对心肌肥大有抑制作用,而Cav3.2对心肌肥大的激活具有重要意义,这2种不同亚型的T型钙通道作用于心肌,对心肌肥大的发生起到调节和平衡作用。
3.2 T型钙通道参与抑制血管过度收缩除心肌细胞外,血管平滑肌细胞亦可表达除Cav1.2外的其他VGCC。钙通道可使血管感知和响应生理刺激,进而调节动脉张力,维持血管功能。其中,T型钙通道受到了广泛关注[35]。Cav3.2在超极化的血管平滑肌细胞局部微结构域信号传递中发挥重要作用,通过调控动脉张力的反馈抑制,引起阻力动脉的扩张[36]。
T型Cav3.2通道介导的Ca2+内流刺激RyR的细胞质结构域,以Ca2+触发的形式促进肌浆网释放Ca2+,引起K+外流和大电导钙激活钾通道(large-conductance calcium-activated potassium channels,BKCa)的激活,从而避免血管过度收缩。为了使这个信号通路正常的发挥作用,T型Cav3.2通道必须与RyR紧密地排列在一起[37]。越来越多研究表明,年龄是影响血管平滑肌功能的关键因素,人们逐渐认识到钙通道信号会随着衰老而改变,而且会影响血管的功能。Fan等[38]利用不同年龄(4和12月龄)小鼠血管平滑肌细胞的高空间分辨率共聚焦Ca2+成像,探究衰老对血管平滑肌细胞中的Cav3.2-RyR-BKCa信号传导通路的影响。该研究表明,衰老破坏了Cav3.2与RyR的偶联,通过蛋白表达分析和电镜观察结果显示,这种作用可能是由于与年龄相关的Cav3.2蛋白表达减少和血管平滑肌细胞微囊结构变化引起的。此外,Georgeon-Chartier等[39]报道了衰老小鼠血管平滑肌细胞RyR介导的Ca2+信号减少,RyR2表达下降,血管收缩能力下降。这些结果表明,衰老可通过破坏Cav3.2-RyR-BKCa信号通路导致血管收缩能力下降。
4 T型钙通道的调节剂
4.1 血管紧张素受体拮抗剂抑制T型钙通道表达众所周知,肾素-血管紧张素系统的激活可以靶向血管平滑肌细胞,从而升高血压,导致高血压,而且该系统同样也与心肌肥大和致死性心律失常有关[40]。该系统的主要分子血管紧张素II已成为影响许多细胞类型和器官功能的关键激素,并在心律失常和心房颤动的发展中起着重要作用[41]。
T型钙通道在心律失常中起重要作用,血管紧张素II 1型(angiotensin II type 1,AT1)受体拮抗剂作为抗心律失常药物发挥作用。心脏的电重构通常与细胞内Ca2+过载有关,T型钙通道能将Ca2+带入心肌细胞,提示T型钙通道在心脏自律性中发挥生理作用。在病理状态下,T型钙通道在心肌细胞中重新表达[42]。心脏电生理特性的变化与包括T型钙通道在内的多种离子通道基因表达的变化有关,这些变化容易导致心律失常的发生[43]。但是T型钙通道如何参与血管紧张素II介导的心律失常的发生,仍需进一步的实验研究。Morishima等[44]研究AT1受体拮抗剂替米沙坦对大鼠新生心肌细胞T型钙通道的表达和心肌收缩力的转录调节的影响。用替米沙坦和血管紧张素II培养心肌细胞24 h后测量T型Ca2+电流及通道。其结果显示,长期应用血管紧张素II(24 h)后,心肌细胞的T型Ca2+电流密度显著增加,并伴有细胞外信号调节激酶1/2(extracellular signal-regulated kinase 1/2,ERK1/2)和p38丝裂原活化蛋白激酶(p38 mitogen-activated protein kinase,p38 MAPK)磷酸化。而使用替米沙坦则降低了Cav3.1和Cav3.2的mRNA表达以及T型Ca2+电流。此外,在缺乏血管紧张素II的情况下,替米沙坦降低了p38 MAPK的基础磷酸化水平,但没有降低ERK1/2的水平。上述结果表明,替米沙坦可通过不依赖激动剂的方式抑制p38 MAPK活性并减弱T型钙通道的表达,从而起到抗心律失常的作用(图1)。
4.2 肾上腺素增强T型钙通道表达心率调节是通过SAN的自动性和传导率的改变发生的,当交感神经受到刺激时,交感神经/β-肾上腺素能系统兴奋,释放儿茶酚胺,与β-肾上腺素能受体结合并激活环腺苷酸(cyclic adenosine monophosphate,cAMP)/蛋白激酶A(protein kinase A,PKA)信号[45]。由于β-肾上腺素能系统对心率调节至关重要,而Cav3.1参与心率产生,因此可以验证β-肾上腺素/PKA系统是否参与对T型钙离子通道的调节。此外,cAMP依赖性PKA对T型钙通道的调控一直存在争议,这可能是由于实验条件、细胞类型的差异以及特定亚型的存在[46]。
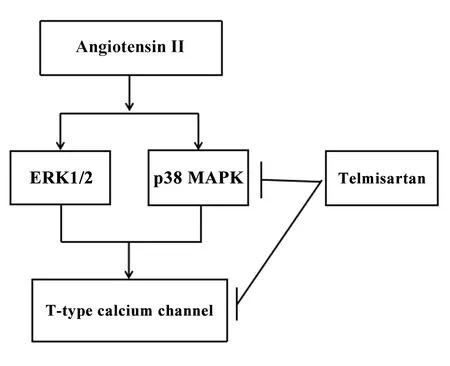
Figure 1.The role of angiotensin and T-type calcium channel.The T-type Ca2+current density in cardiomyocytes was increased after angiotensin II treatment via activating extracellular signal-regulated kinase 1/2(ERK1/2)and p38 mitogen-activated protein kinase(p38 MAPK).In contrast,telmisartan decreased the density of T-type Ca2+current in a dose-dependent manner by inhibiting the basal phosphorylation level of p38 MAPK in the absence of angiotensin II.图1 血管紧张素与T型钙通道的作用
Li等[47]通过Cav3.1双转基因小鼠心室肌细胞,以及野生型小鼠、Cav3.1敲除小鼠和Cav3.2敲除小鼠SAN细胞的研究,观察到β-肾上腺素能受体激动剂异丙肾上腺素能够增强过表达Cav3.1通道的活性。这种作用是由腺苷酸环化酶(adenylate cyclase,AC)/cAMP/PKA系统介导的,因为cAMP能够表现异丙肾上腺素的作用效果,而PKA抑制剂H89可以消除异丙肾上腺素对Cav3.1的作用。由此说明cAMP/PKA通路介导β-肾上腺素能受体刺激而增强Cav3.1活性。之后,Li等[48]提出β-肾上腺素能受体通过刺激T型钙通道活性上调,诱导心率升高和房室传导加速。对小鼠注射异丙肾上腺素,其EC50在Cav3.1转基因小鼠的心率应答中较在Cav3.1基因敲除小鼠中低。而在异丙肾上腺素使小鼠心率应答的占比中,Cav3.1转基因小鼠高于Cav3.1基因敲除小鼠。而且Cav3.1基因敲除小鼠的T型Ca2+电流消失,Cav3.1转基因小鼠中的T型Ca2+电流增强。这说明小鼠SAN中的T型Ca2+电流是由Cav3.1介导的,β-肾上腺素能受体刺激心率上调和房室传导加速的作用在KO小鼠体内减弱,而在Cav3.1转基因小鼠体内加强;Cav3.1的表达影响SAN细胞自律性,而且其表达通过β-肾上腺素能受体刺激调节。这些研究说明β-肾上腺素能受体刺激Cav3.1活性增强在小鼠心率调节中发挥重要作用,β-肾上腺素能系统调控Cav3.1异常可能会导致心脏自律性异常(图2)。
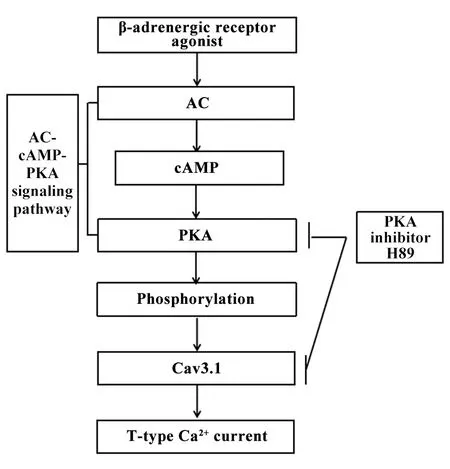
Figure 2.The role of adrenaline and T-type calcium channel.βadrenergic receptor agonist increased the expression of Cav3.1 by activating adenylate cyclase(AC)-cyclic adenosine monophosphate(cAMP)-protein kinase A(PKA)signaling pathway,and H89,a PKA inhibitor,could inhibit the phosphorylation of PKA and reduced the T-type Ca2+current.图2 肾上腺素与T型钙通道的作用
4.3 醛固酮间接调控T型钙通道表达盐皮质激素,尤其是醛固酮,与心肌肥大和心力衰竭的发生密切相关。心脏中盐皮质激素受体(mineralocorticoid receptor,MR)的激活是心血管疾病发生的危险因素。MR激活会导致包括钙通道在内的许多离子通道的上调,发生心脏肥大和心律失常[49]。Messaoudi等[50]对心肌细胞靶向性MR过度表达的小鼠及其对照组给予1周低剂量醛固酮治疗,确定醛固酮是否能在体内激活心肌细胞MR。通过分析心脏转录组,观察到心肌细胞中存在醛固酮调节的基因,并激活心肌细胞MR。由于T型钙通道内不包含任何盐皮质激素反应元件(mineralocorticoid response element,MRE;与MR锚定的结构域一致的核苷酸序列),醛固酮对T型钙通道表达的控制可能是间接的[51],可能是由于心肌细胞MR替代了MRE的作用。
心血管疾病与微小RNA(microRNA,miRNA,miR)的调节密切相关[52]。miRNA调节大量信号通路,参与细胞间的信息交流,并参与心血管系统的生理病理进程,如调控心肌细胞分化、促进血管新生、维持内皮细胞稳定性及抑制内皮迁移和增殖等。Rossier[53]提出miR-204在MR下游发挥作用以控制T型钙通道表达。Koyama等[51]假定一种特定的miRNA在MR激活与T型钙通道表达和心肌细胞搏动频率的调节中起到联系作用。通过一项筛选实验发现,在醛固酮刺激离体乳鼠心肌细胞后,miR-204是主要上调的miRNA之一。当miR-204在分离的心肌细胞中过度表达时,其自发搏动频率在24 h后显著增加,与受到醛固酮刺激时表现相同,编码T型钙通道的mRNA增加。同时,miR-204过度表达后,T型Ca2+电流显著增加。当抑制miR-204的表达时,会消除醛固酮诱导Cav3.1和Cav3.2的mRNA表达增加,同时T型Ca2+电流也会减少。此外,Koyama等[51]还观察到醛固酮和miR-204的过度表达降低了已知的Cav3转录抑制因子——神经元限制性沉默因子(neuron-restrictive silence factor,NRSF)。由此可以说明,醛固酮对T型钙通道的调控是间接实现的,醛固酮对心肌细胞MR的刺激会导致miR-204的表达,而miR-204的表达促进了分离的大鼠心室肌细胞中T型钙通道的表达,增加了细胞自发收缩的频率,这是通过抑制NRSF蛋白实现的(图3)。

Figure 3.The role of mineralocorticoid and T-type calcium channel.The stimulation of mineralocorticoid receptor(MR)by aldosterone could increase T-type Ca2+current via microRNA-204(miR-204).Neuron-restrictive silencing factor(NRSF)inhibited this pathway by lessening the level of miR-204 and T-Type calcium channel.图3 盐皮质激素与T型钙通道的作用
5 结语
VGCC不仅能够改变细胞电位,还能参与信号转导。不同于其它高电压激活的钙通道,T型钙通道以其低电压激活的特性在心脏血管的生理活动中发挥着重要的作用,参与心脏自律性的产生和传导,其功能缺陷可导致先天性心脏传导阻滞的发生。不同亚型的T型钙通道对心肌肥大作用不同作用。除心肌本身外,T型钙通道还可参与抑制血管过度收缩,进而维持正常血管收缩舒张功能。近年来对于T型钙通道在电生理传导通路作用及分子调控机制仍是研究热点问题,虽然已证实其受肾上腺素、醛固酮等多种激素的直接或间接调控,但其他调控因素及相应的临床转化应用仍需进一步的探索。
猜你喜欢
世界最新医学信息文摘(2020年68期)2020-12-25
小学科学(学生版)(2019年10期)2019-11-16
中国外汇(2019年23期)2019-05-25
延安大学学报(医学科学版)(2019年1期)2019-03-29
中国环境监察(2017年5期)2017-10-23
中国当代医药(2015年30期)2015-03-01
中西医结合心血管病杂志(电子版)(2015年35期)2015-01-21
中国药理学通报(2013年11期)2013-12-08
中国医学科学院学报(2013年6期)2013-03-11
中国烟草学报(2012年5期)2012-04-12
