
铒掺杂羟基磷灰石的荧光及其生物矿化性能
2022-12-06 06:29白春华侯欢健杨小宁李光辉
无机化学学报 2022年11期
白春华 侯欢健,2 杨小宁 李光辉*,
(1内蒙古科技大学矿业与煤炭学院,包头 014010)
(2中钢马鞍山矿山研究总院有限公司,马鞍山 243000)
离子掺杂的羟基磷灰石(hydroxyapatite,HAP)颗粒是一种具有良好生物相容性的矿物,其性能因制备路线、前驱体的影响而有差异,不同离子取代也对其结晶度、微晶尺寸、晶格参数、形貌、生物活性和荧光性能有一定影响[1-3]。稀土离子具有发光强度大、多色可调(可见光-近红外光区)、单色性好、发光寿命长的光学特性,因此,将稀土离子与羟基磷灰石两者的优越性能结合具有重要的研究价值[4-6]。一方面,稀土是一种非生物必需元素,近年来,随着稀土矿业的持续开发及含稀土产品的广泛应用,稀土元素尤其是水溶性较好的稀土离子(如Er3+等)会逐渐分散至矿区及扩散到城市的土壤、水、空气环境中,进而进入生物链系统,且这种全球化扩散的趋势还会得到加强。早期研究发现,人体各器官对稀土的吸收主要积聚于骨骼中,并随着年龄增长而不断累积[7-9],这要求对稀土离子在骨骼中的赋存状态即稀土离子嵌入磷灰石矿物中的结合状态进行基础研究,以便了解骨矿物相中稀土离子是否会对人们的长期健康构成潜在影响以及影响的机制。而另一方面,稀土离子在医学领域应用也有不可替代的优良性能[10-11],如La3+取代的HAP能抑制龋齿扩散,Ce3+掺杂的HAP可以提高生物降解性和抗菌性能,169Er(t1/2=9.5 d,β射线)用于放射性关节炎小关节滑膜切除术治疗。Esam等用Er2O3作为铒源掺杂磷酸钙,为激光能量的吸收提供光场所,使人造釉质矿物在牙本质上更加致密[12]。Alshemary等用微波辅助沉淀法合成了Er-HAP,Er3+的荧光特性可用于研究Er-HAP在体内的吸收状况,并用来进一步监测患者的骨愈合过程[2]。随着体内成像技术的发展,稀土离子掺杂纳米材料的光致发光光谱检测已越来越多地应用于光动力疗法和药物递送等医疗领域[13-15]。
我们采用共沉淀法合成了Er掺杂量(物质的量分数)分别为1%、9%的Er-HAP微球状颗粒,探究了不同掺杂量对Er-HAP的晶体结构、表面化学成分和发光特性的影响,并通过模拟体液(simulated body fluid,SBF)浸渍实验进一步分析了其生物矿化性能,这一研究为Er-HAP颗粒在生物医学成像领域的潜在应用提供基础,也为Er3+嵌入骨骼中与生物磷灰石的赋存状态研究提供参考。
1 实验部分
1.1 试剂
四水合硝酸钙((Ca(NO3)2·4H2O)、磷酸氢二铵((NH4)2HPO4)、六水合硝酸铒((Er(NO3)2·6H2O)和氢氧化钠(NaOH)均为市售分析纯,水为市售纯净水。
1.2 仪 器
Er-HAP颗粒的X射线光电子能谱(XPS)通过X射线光电子能谱仪(ESCALAB 250Ⅺ,美国赛默飞-世尔公司)获得。形貌与能量色散X射线谱(EDS)分析由场发射扫描电子显微镜(SEM,S-4800,日本日立公司)获得,电子枪为冷场发射电子源,工作电压为20 kV。通过X射线衍射仪(XRD,D8 ADVANCE,德国布鲁克公司)检测晶体结构,辐射源为Cu靶Kα射线,波长为0.154 6 nm,2θ扫描范围为20°~70°和5°~60°,扫描速度为 4(°)·min-1,光管电压为 40 kV,光管电流为40 mA。通过UV/Vis/NIR分光光度计(UH-4150,日立高新技术公司)对样品的紫外可见漫反射(UV-Vis DRS)光谱进行了扫描。荧光光谱通过稳态瞬态荧光光谱仪(FLS1000/FS5,英国爱丁堡公司)获得。
1.3 实验过程
采用化学共沉淀法制备了不同Er3+含量的Er-HAP颗粒。实验操作过程如下:在搅拌速率500 r·min-1和 室 温 下 ,依 次 称 取 Ca(NO3)2·4H2O 和Er(NO3)2·6H2O(分别为 0.099和0.001 mol),加入三口烧瓶(500 mL)中,然后加入30 mL水溶解。再称取(NH4)2HPO4(0.06 mol)溶于30 mL水中,将(NH4)2HPO4溶液缓慢滴加到上述混合溶液中,控制滴加时间约为30 min。然后用饱和NaOH溶液将反应液pH值调至12.20,再于120℃搅拌加热10 min,最后将混合溶液陈化12 h。陈化完成后,抽滤,得到的浅粉色沉淀物经纯净水洗涤(30 mL×3),80℃下真空干燥4 h。干燥后的样品用玛瑙研钵研磨得到浅粉色粉末,将样品放入马弗炉中煅烧(800℃,2 h),冷却后的样品密封,用于测试与实验。样品记为Er(1%)-HAP。将Ca(NO3)2·4H2O和Er(NO3)2·6H2O的加入量改为0.091和0.009 mol,按上述过程制备,得到样品Er(9%)-HAP。
SBF浸泡实验:SBF按照标准配方配制(表1)[16]。称取0.2 g的Er-HAP置于50 mL配好的SBF溶液中,在37℃恒温水浴中浸渍72 h,浸泡完成后将反应液进行离心分离,所得固体常压干燥(80℃,3 h)后密封备用。
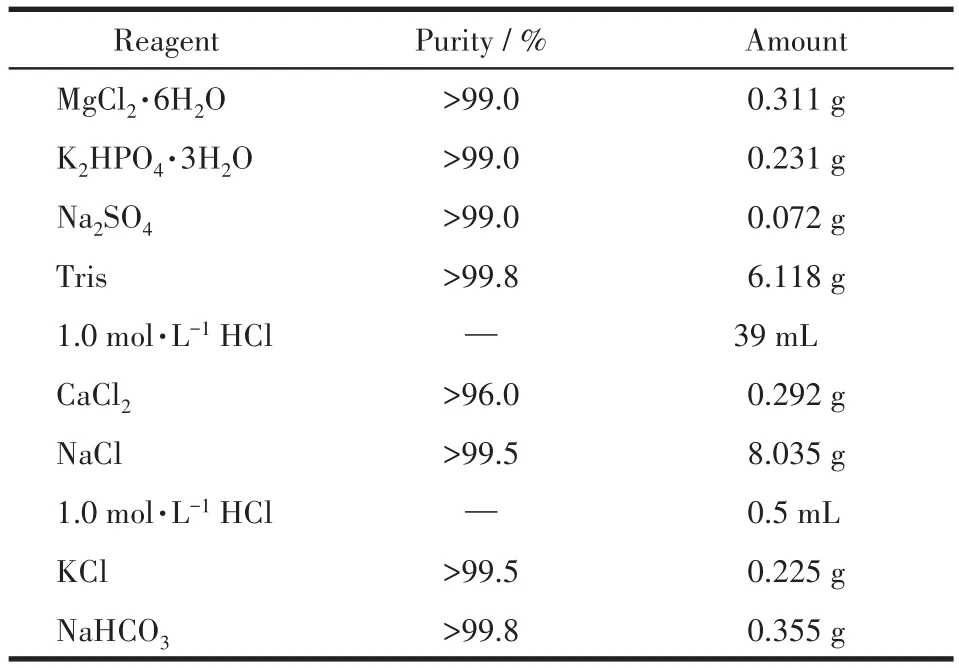
表1 1 000 mL SBF的配方Table 1 Recipe for 1 000 mL SBF
1.4 理论模拟
除了XRD测试以外,还应用Materials Studio软件中的Reflex模块对Er(9%)-HAP颗粒的晶体结构进行了模拟计算。
2 结果与讨论
2.1 Materials Studio模拟与XRD分析
HAP(Ca10(OH)2(PO4)6)的空间群为六方晶系的P63/m,a=b=0.941 80 nm,c=0.688 40 nm。应用Materials Studio软件构建Er(9%)-HAP的晶体结构模型(图 1),可观察到 HAP 结构中有 4 个 Ca2+(Ca1),分别占据Z=0和Z=1/2的位置,另外6个Ca2+(Ca2)中的3个位于Z=1/4的位置,余下3个位于Z=3/4的位置,掺杂后,其中一个被Er替代。表2为Rietveld法精修计算的Er(9%)-HAP的晶胞参数,表中数据显示,相对于纯的HAP,其a轴(均值为0.906 72 nm)被压缩,且沿c轴(均值为0.717 46 nm)择优取向生长,这说明Er3+掺杂使HAP晶体生长在c轴方向上有择优取向能力。
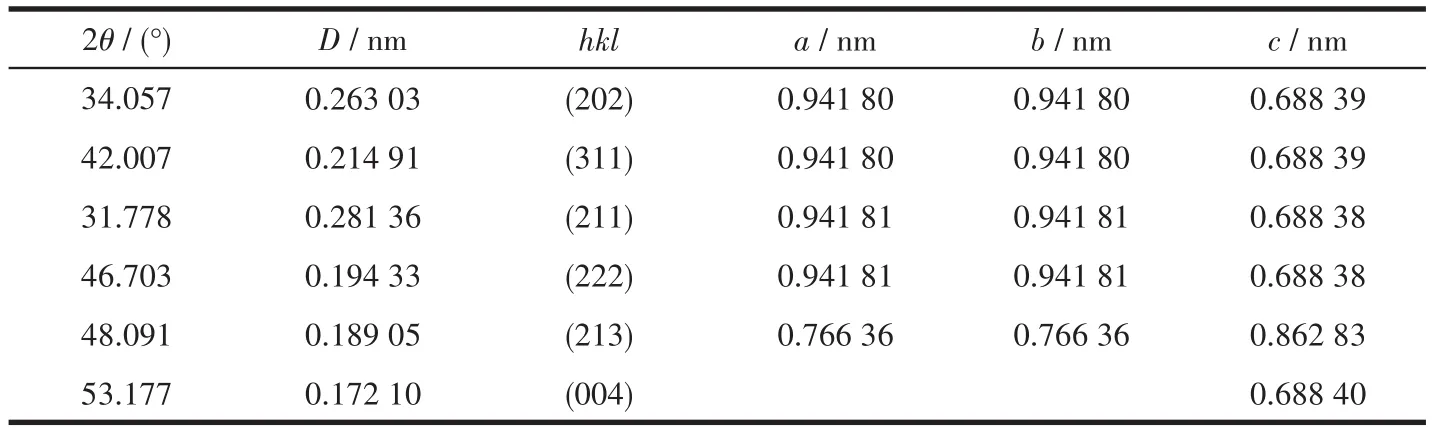
表2 Er(9%)-HAP颗粒的晶胞参数Table 2 Unit-cell parameters of Er(9%)-HAP particles
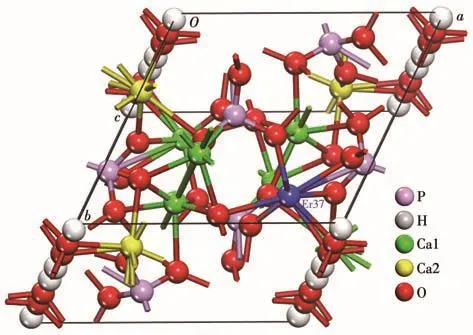
图1 Er(9%)-HAP的晶体结构图Fig.1 Crystal structure of Er(9%)-HAP
图2为Er(9%)-HAP颗粒的XRD图,可以观察到应用Materials Studio软件模拟出它的各个晶面衍射峰(图2a)与实验测出的衍射峰(图2b)位置基本保持一致,与Er3+替代了HAP晶格中Ca2中的一个Ca2+位点的观点相符。进一步与HAP的PDF标准卡片(No.09-0432)比对发现,在 2θ为25.9°、28.9°、31.8°、32.9°、34.1°、39.8°、46.7°和 49.5°位置处的 8个主峰分别对应于羟基磷灰石的(002)、(210)、(211)、(300)、(202)、(310)、(222)和(213)晶面[17]。此外,还匹配到了(100)、(101)、(110)、(200)、(111)、(102)、(211)、(301)、(212)、(221)、(311)、(113)、(203)、(312)、(320)、(321)、(410)、(402)、(004)、(322)和(313)等多个次峰,共计 29个晶面,且样品中没有匹配到属于Er2O3的衍射峰,表明Er3+替代了Ca2+,成功掺杂到HAP的晶体结构中[2]。
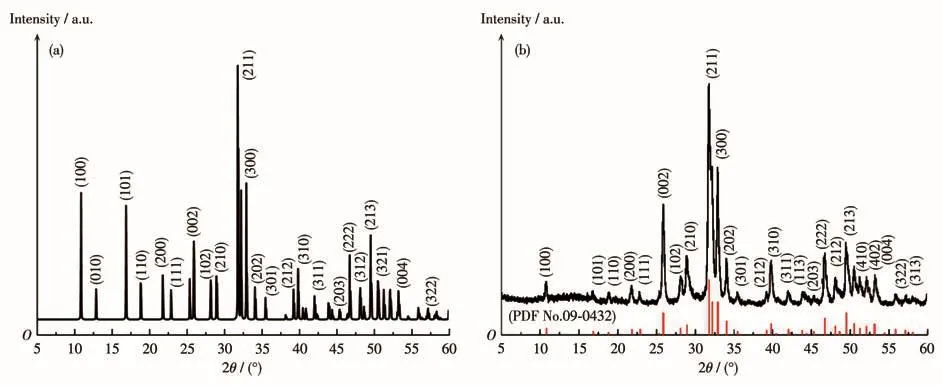
图2 Er(9%)-HAP颗粒的模拟(a)和实验(b)XRD图Fig.2 Simulated(a)and experimental(b)XRD patterns of Er(9%)-HAP particles
根据衍射峰的相对强度变化可以推测出晶体不同晶面的生长速度,对比各衍射峰的强度可以发现,Er(9%)-HAP颗粒的I(211)∶I(002)=1∶0.40,而HAP标准卡片的I(211)∶I(002)=1∶0.47,(002)晶面垂直于c轴,其相对强度显著增强,意味着Er(9%)-HAP晶体有沿c轴增长的趋势,(300)和(310)所代表的a轴变化不大,Er(9%)-HAP的c/a值明显增大。从理论上来说,较小尺寸的Er3+(0.088 nm)取代较大尺寸的Ca2+(0.099 nm),会导致晶胞参数和晶面间距变小,衍射峰向更高衍射角方向移动,这一偏移与XRD测试的实验结果一致,进一步印证了Er3+已掺杂到HAP的晶格中[18]。
2.2 SEM和EDS分析
图3展示了Er(9%)-HAP颗粒的SEM图。从图中可以清楚地观察到其形貌呈微球状,绝大多数颗粒的粒径小于500 nm,且颗粒分布较为均匀,由于羟基磷灰石具有较高的表面能,因而能观察到几个或数十个微球聚集成微球团簇的现象[19]。与纯HAP的微球状相比[18],掺杂Er3+的HAP颗粒大多数呈椭球状(图3中黄色方框部分尤为明显),其形貌不是理想的球状,而是沿着某一径向有伸长,表明Er3+掺杂对HAP的形貌有一致性的影响,这与XRD的表征结果一致:(210)晶面强度增大,Er-HAP沿着c轴增长,c/a增大,也与Materials Studio模拟的结果一致。
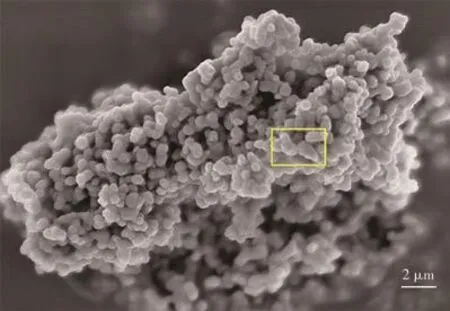
图3 Er(9%)-HAP颗粒的SEM图Fig.3 SEM images of Er(9%)-HAP particles
从Er(9%)-HAP颗粒的EDS面扫图(图4d)可以清楚地观察到样品中Er元素的存在,且分布较为均匀,说明Er3+成功掺杂到HAP晶格中。元素含量结果(表3)显示Er(9%)-HAP颗粒的Ca与P的物质的量之比(Ca/P比)为1.17,远低于HAP(1.66),其中原因一部分是由于Er3+取代了Ca2+进入到HAP晶格中[20],另一部分则来自于分析方法的差异,由于EDS是一种表面分析手段,而Er-HAP颗粒表面的Ca/P比会随表面的相变而发生改变[21],类似的改变在磷灰石矿物的表面结构中经常被检测到[22-23]。
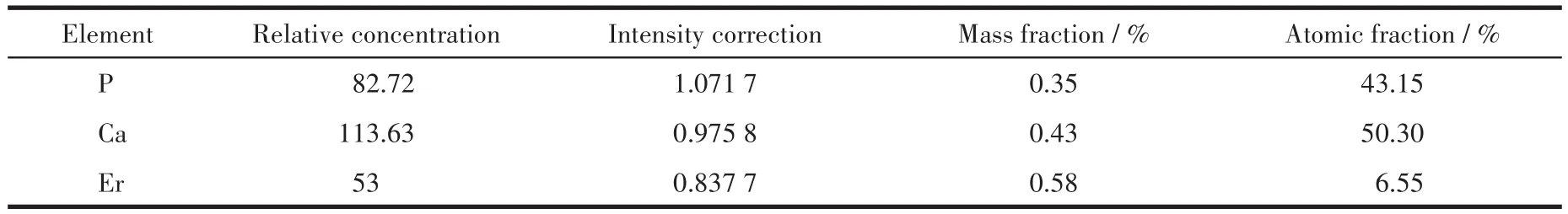
表3 Er(9%)-HAP颗粒表面的元素含量Table 3 Contents of the elements of the surface of Er(9%)-HAP particles

图4 Er(9%)-HAP颗粒的EDS面扫图Fig.4 EDS mappings of Er(9%)-HAP particles
此外,EDS谱图显示Er(9%)-HAP颗粒的Ca和Er的物质的量之比约为7.68,而实验中投料时两者的物质的量之比应为9,意味着Er-HAP颗粒表面的Er元素含量偏高,表明当溶液中有化学性质相似的稀土金属离子Er3+与碱土金属离子Ca2+同时存在时,在生成羟基磷灰石沉淀的过程中,离子半径稍小的Er3+会竞争性地优先占据羟基磷灰石的晶格,从而将少部分Ca2+排挤到水中,这与稀土离子易在人体骨骼中蓄积的事实相符。
2.3 XPS分析
图5为Er(9%)-HAP颗粒的XPS谱图。从XPS全谱图(图5a)中可以清楚地观察到Ca、O、P、C和Er五种主要元素的存在,其中 O1s、Ca2p、Ca2s、Ca3p、P2s和P2p峰位都比较明显。对XPS谱图中的C、Ca、O、P、Er原子的峰位进一步分峰拟合,C1s高分辨谱可以分出3个峰(图5b),其中在284.8 eV处的峰归属于外源性的污染碳元素,在285.8 eV处的峰归属于C—O中的C元素,在288.4 eV处的峰归属于CO32-,两者均可能来自于沉淀反应过程中从空气中吸收的CO2[22];而Ca2p高分辨谱可以分出2个峰(图5c),其半高宽近似相等,分别为1.50和1.49,350.4和346.9 eV处的峰相应地归属于Ca—O—P的Ca2p1/2以及Ca2p3/2轨道[25-26];进一步,P2p高分辨谱可以分出4个峰(图5d),在132.3 eV处的峰归属于P—O—H单元,133.6和132.7 eV的2个峰半高宽均为1.7,结合能相差0.9 eV,面积比为1∶2,分别归属于PO43-中磷原子的2p1/2、2p3/2轨道,在133.9 eV处的峰归属于P=O键[27-28];O1s高分辨谱可以分出4个峰(图5e),其中在530.2 eV的峰归属于CO32-中的O元素,在530.6 eV的峰归属于P—O中的O元素,在531.3 eV的峰归属于P=O中的O元素,在531.8 eV的峰归属于O—H中的O元素[29];由于Er元素的信号较弱,仅在168.9 eV处观察到一个Er4d的峰位(图5f),表明Er3+成功掺杂到了羟基磷灰石晶格中,与XRD、EDS结果一致。
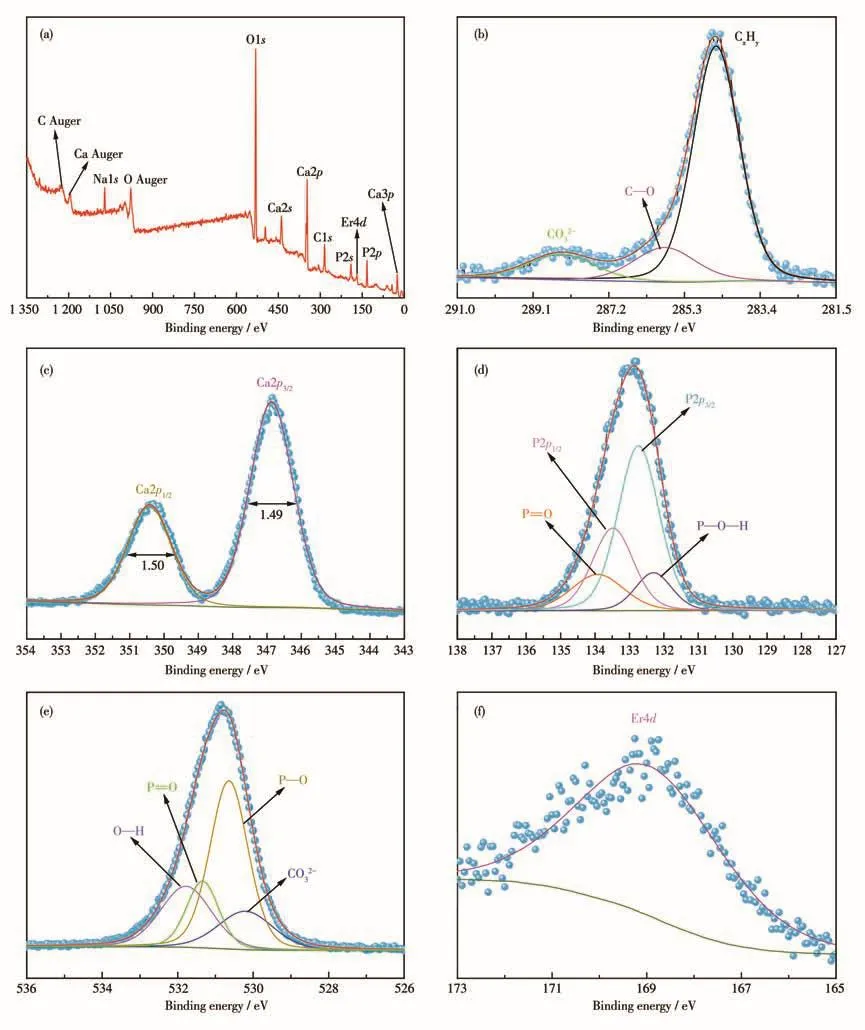
图5 Er(9%)-HAP颗粒的XPS谱图:(a)全谱;(b)C1s;(c)Ca2p;(d)P2p;(e)O1s;(f)Er4dFig.5 XPS spectra of Er(9%)-HAP particles:(a)survey;(b)C1s;(c)Ca2p;(d)P2p;(e)O1s;(f)Er4d
2.4 UV-Vis DRS光谱与荧光光谱分析
图6显示了Er(9%)-HAP在320~1 100 nm波长范围内的UV-Vis DRS光谱,可以清楚地观察到分别位于 365、375、405、449、489、520、541、654、802 和975 nm处的10个吸收峰,它们依次对应Er3+从基态4I15/2能级跃迁到4G9/2、4G11/2、2H9/2、4F5/2、4F7/2、2H11/2、4S3/2、4F9/2、4I9/2和4I11/2能级[2],且从4I15/2→2H11/2能级的跃迁强度最高,这一超敏感跃迁(hypersensitive transition)[30]表明Er(9%)-HAP颗粒有光致发光性能。
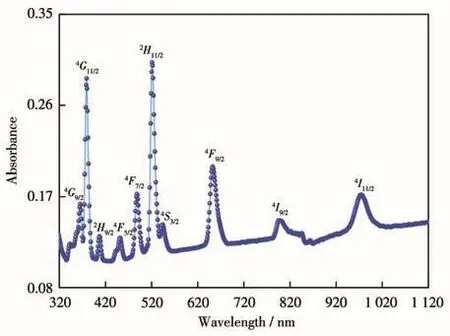
图6 Er(9%)-HAP颗粒的UV-Vis DRS谱图Fig.6 UV-Vis DRS spectrum of Er(9%)-HAP particles
图7为Er(9%)-HAP颗粒的发光衰减拟合曲线。通过对其荧光寿命数据进行拟合,得到二阶的指数衰减曲线,其公式[31]为 It=A1exp(-t/τ1)+A2exp(-t/τ2)+y0,其中,It是时间为 t时的发射强度,A1和 A2是常数,τ1和τ2代表了发光寿命的2个相关值,τ1和τ2的值可以由方程拟合计算得到,拟合结果如表4所示。再根据公式[32]:τ=(A1τ12+A2τ22)/(A1τ1+A2τ2),将拟合后的数据代入公式,对样品的发光寿命进行计算,得到平均衰减时间τ=2.731 4 ns。这个结果表明,Er(9%)-HAP颗粒有较短的荧光寿命,但其发光强度则有明显优势[33],基于HAP良好的生物相容性,因此可以乐观地认为其在生物医学影像领域有着良好的应用潜力。
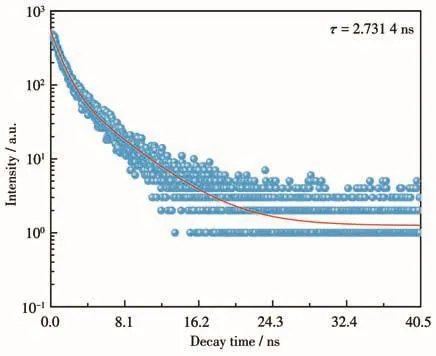
图7 Er(9%)-HAP颗粒的荧光寿命拟合图Fig.7 Fitting diagram for fluorescence lifetime of Er(9%)-HAP particles
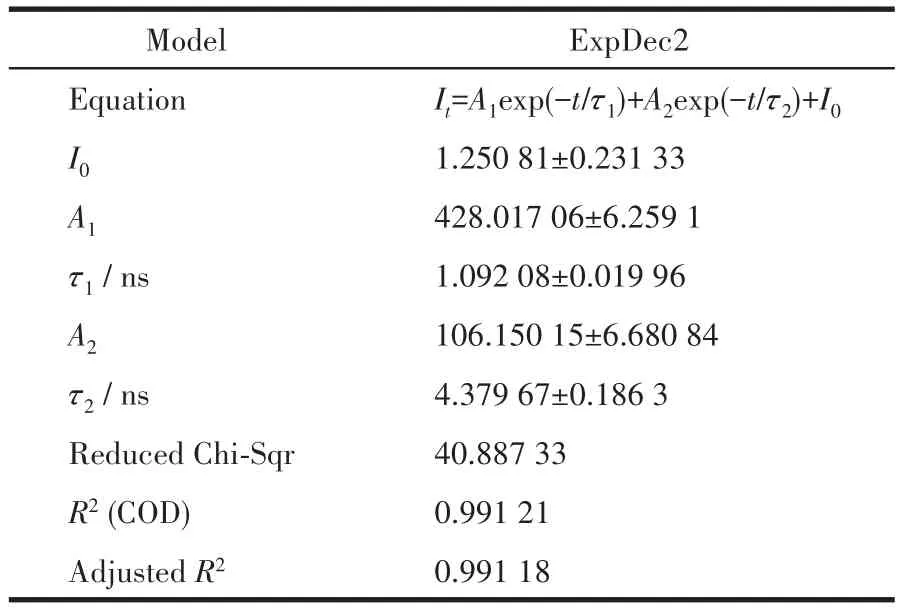
表4 Er(9%)-HAP颗粒的荧光寿命拟合数据Table 4 Fluorescence lifetime fitting data of Er(9%)-HAP particles
图8a为Er(9%)-HAP颗粒的荧光发射光谱。当激发波长为340 nm,Er-HAP颗粒的发射光谱显示出4个相应的发射带:紫色(419 nm)、蓝色(458 nm)、绿色(501 nm)和绿色(535 nm),分别对应4F3/2→4I15/2、4F5/2→4I15/2、4F7/2→4I15/2、4S3/2→4I15/2上的转换[2,20]。图 8b阐释了Er(9%)-HAP颗粒在受激发射过程中涉及的不同机制。Er3+的基态处于低能电子的4I15/2态,当其被340 nm的紫外光激发后,低能电子被光子激发跃迁到处于高能级的4G11/2态。激发态必然变得不稳定,可以有2种方式来释放能量:大部分受激发的Er3+将通过与邻近其他原子(包括Ca2+、Er3+)发生碰撞来释放出多余的这部分能量,少部分受激发的Er3+则直接以光的形式来释放出多余的这部分能量,即发生非辐射衰减(nonradiative decay,NR),形成光致发光。在碰撞过程中,颗粒表面的Er3+会被优先激发,而受激发Er3+中的一部分会碰撞与其相邻的且位于表面稍内侧的未能受到紫外光激发的Er3+离子,从而将其能量转移到这些Er3+离子上,进而导致跃迁到高能态的Er3+浓度增加,引起光致发光效应的增强[34]。
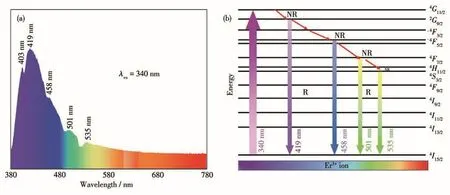
图8 Er(9%)-HAP颗粒的(a)荧光光谱及其(b)能级示意图Fig.8 (a)Fluorescence spectrum and(b)schematic energy levels of Er(9%)-HAP particles
2.5 SBF浸泡反应前后的SEM和EDS比较分析
图9展示了Er(9%)-HAP颗粒在SBF中浸泡72 h后的SEM和EDS面扫图。相比于图3,浸泡后的矿物表面(图9a)新形成了一层完全不同于原始表面结构的沉积层,呈发育尚不完善的不规则片状晶体,与文献中呈叶片状晶体的矿化层相似[35]。结合EDS面扫图(图9b~9d)可以观察到形成的这些矿化层属于磷酸钙型矿物,且新形成的矿化层处的Er3+分布相对较稀薄,而无新形成矿化层的区域,其Er3+分布相对较密,这可能来自于两方面因素的联合影响:一是位于表面的部分Er3+与SBF中的阳离子发生了离子交换反应,浸泡后,Er(9%)-HAP颗粒的(nEr+nCa)/nP由初始的1.32增加至1.63(表5),其与HAP的Ca、P化学计量比1.67更为接近,可以看出较多Ca2+沉积在Er-HAP表面,Er3+占比明显减少,从而导致Er3+含量下降;另一个因素是由于在那些被新矿化层所覆盖的区域,能被探测到的表面Er3+信号下降,形成的矿化层在Ca、P化学计量比与形貌两方面均接近HAP。
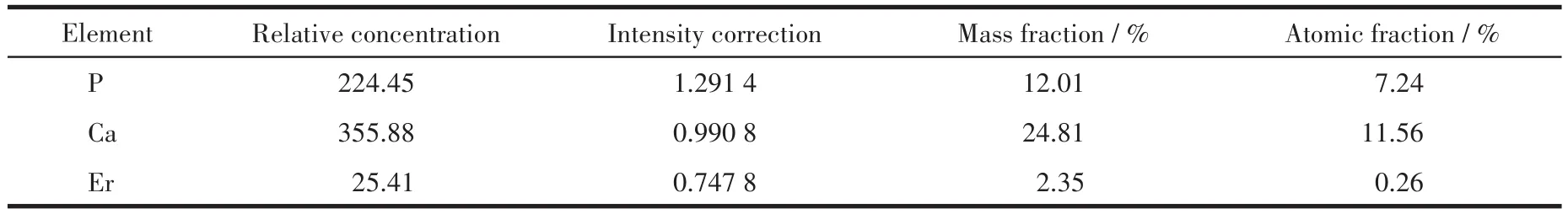
表5 Er(9%)-HAP在SBF中浸泡72 h后颗粒表面的元素含量Table 5 Contents of the elements of the surface of Er(9%)-HAP particles after being immersed in SBF for 72 h

图9 Er(9%)-HAP颗粒在SBF中浸泡72 h后的(a)SEM图和(b~d)EDS面扫图Fig.9(a)SEM image and(b-d)EDS mappings of Er(1%)-HAP particles after being immersed in SBF for 72 h
图10和图11分别为Er(1%)-HAP颗粒在SBF中浸泡72 h前后的SEM和EDS面扫图。相比于浸泡前的形貌(图10a),浸泡后Er(1%)-HAP颗粒的表面出现了不规则超薄片状的矿化沉积物(图10a),能观察到矿物片站立于颗粒表面时其因超薄而带来的扭曲和旋转特征,这些超薄片相互交织围成较致密的网状通道,表明其矿化速度相比Er(9%)-HAP颗粒表面的矿化速度明显增大。结合EDS面扫图(图11b~11d)可以观察到 Ca、P 和 Er元素均匀分散在Er(1%)-HAP颗粒中。SBF浸泡后,Er(1%)-HAP颗粒的(nEr+nCa)/nP基本保持不变(由1.37稍降至1.30)(表6和7),与磷酸八钙的理论Ca、P化学计量比1.33更接近,这表明当Er元素含量较低时,其形成的矿化层在Ca、P化学计量比与形貌两方面更接近于磷酸八钙的相应特性。
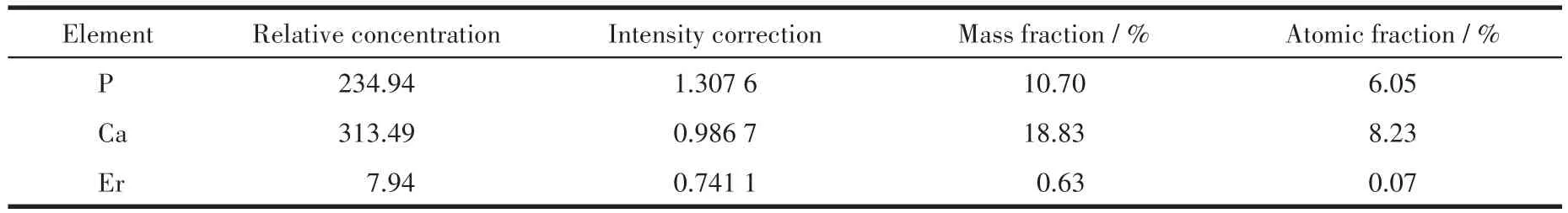
表6 Er(1%)-HAP颗粒表面的元素含量Table 6 Contents of the elements of the surface of Er(1%)-HAP particles

图10 Er(1%)-HAP颗粒的(a)SEM图和(b~d)EDS面扫图Fig.10 (a)SEM image and(b-d)EDS mappings of Er(1%)-HAP particles

图11 Er(1%)-HAP颗粒在SBF中浸泡72 h后的(a)SEM图和(b~d)EDS面扫图Fig.11 (a)SEM image and(b-d)EDS mappings of Er(1%)-HAP particles after being immersed in SBF for 72 h
研究表明,相比于HAP,磷酸八钙有更优的成骨性能[36-38]。通过比较2种Er-HAP颗粒分别在SBF浸泡反应前后的SEM和EDS面扫图发现:(1)不同Er3+掺杂量(xEr3+≤9%)的Er-HAP在SBF中浸泡均能形成矿化层,初步表明Er-HAP颗粒都具有生物矿化能力[39]。(2)Er3+掺杂量对Er-HAP表面的生物矿化性能有负面影响。一是在矿化速率上,相比Er3+掺杂量1%的Er-HAP,随着HAP中Er3+掺杂量的显著增加,表面矿化速度显著下降,表明Er3+掺杂会抑制HAP颗粒表面矿化层的形成速率;二是形成的矿化层特性也不同,当Er掺杂量在1%左右时,形成的矿化层为超薄片状,更接近磷酸八钙的特性,随着HAP中Er3+掺杂量的显著增加,矿化层由超薄片状转变为叶片状,更接近HAP的特性,也就意味着其成骨性能显著下降。总之,随着Er3+掺杂量的显著提高,Er-HAP颗粒表面的矿化速度和成骨性能均明显下降。基于稀土离子与钙离子之间理化性质的相似性,本实验结果还提供了一个更广泛的猜想,即如果长期生活在稀土矿区或工业区,当摄入稀土离子污染的食物与饮用水导致骨骼中累积的稀土含量超过某一阈值时,成骨性能和骨质量可能会出现下降的倾向。不过稀土元素含量升高对骨骼生理、生物化学、形态及骨质量的持续影响需要更长期深入的科学研究。最后,综合考虑到应用于生物医学成像时Er-HAP颗粒所需的结晶性、荧光性能,我们认为Er掺杂量在1%左右时,能获得较优的结晶性、荧光性能与生物矿化能力的组合。
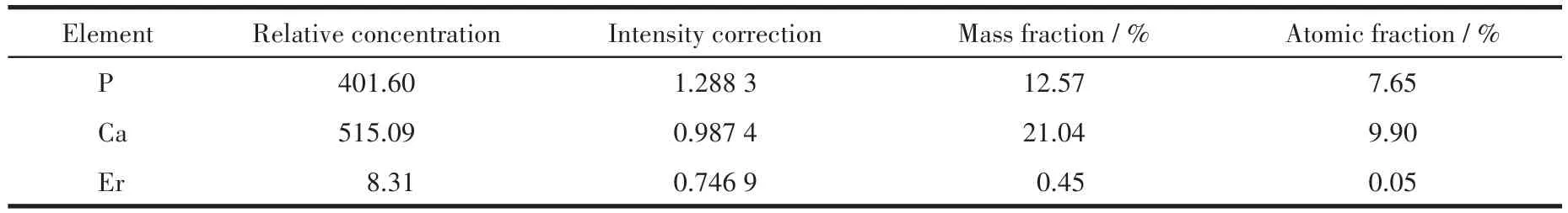
表7 Er(1%)-HAP颗粒在SBF中浸泡72 h后颗粒表面的元素含量Table 7 Contents of the elements of the surface of Er(1%)-HAP particles after being immersed in SBF for 72 h
3 结 论
(1)采用化学共沉淀法合成了光活性的Er-HAP颗粒。微球状的Er-HAP颗粒有高的结晶度,除Er3+替代了HAP晶格中Ca2位置中的一个Ca2+位点以外,其晶体结构与羟基磷灰石基本相同。这表明,当溶液中Er3+与Ca2+同时存在时,在生成羟基磷灰石沉淀的过程中,离子半径稍小的Er3+会竞争性地占据羟基磷灰石的晶格。
(2)Er-HAP颗粒在受到波长340 nm的紫外光激发后具有光致发光性能,显示了4个发光带,分别归属 于4F3/2→4I15/2、4F5/2→4I15/2、4F7/2→4I15/2、4S3/2→4I15/2跃迁,基于HAP良好的生物相容性,表明Er-HAP颗粒有成为一种生物医学成像材料的潜力。
(3)随着Er3+掺杂量的显著提高,Er-HAP颗粒表面的矿化速度和成骨性能均显著下降。综合考虑到Er-HAP颗粒应用于生物医学影像领域时所需要的适当结晶性与荧光性能,我们认为Er掺杂量在1%左右时,该颗粒能获得较强的生物矿化能力。
猜你喜欢
建材发展导向(2022年24期)2022-12-22
岩石学报(2022年10期)2022-11-12
环境卫生工程(2021年4期)2021-10-13
能源工程(2021年3期)2021-08-05
昆明医科大学学报(2021年1期)2021-02-07
矿产综合利用(2020年1期)2020-07-24
中成药(2018年2期)2018-05-09
中国塑料(2016年8期)2016-06-27
中国塑料(2016年12期)2016-06-15
人间(2015年11期)2016-01-09
