
Expression and Purification of Serine/Arginine-Rich Splicing Factor 1 from Escherichia coli Expression System
2022-12-09 14:22ZHANGMinmin张敏敏ZHANGYunlong张云龙CHENTingLUChangrui陆昌瑞
ZHANG Minmin(张敏敏), ZHANG Yunlong(张云龙), CHEN Ting(陈 婷), LU Changrui(陆昌瑞)
College of Biological Science and Medical Engineering, Donghua University, Shanghai 201620, China
Abstract: Serine/arginine-rich splicing factor 1 (SRSF1), as a prototype member of the highly conserved serine/arginine family of RNA binding proteins, plays an important role in mRNA alternative splicing, stabilization, nuclear export, and translation. Here, the expression system was established to purify full-length human SRSF1 from Escherichia coli (E. coli). The SRSF1 coding sequence was amplified by polymerase chain reaction (PCR) and inserted into the pET-28a-ppSUMO vector with His-tag to construct a recombinant plasmid His-SUMO-SRSF1. Then the plasmid was transformed into BL21 (DE3) competent cells for expression. After purification by affinity chromatography and cleavage of His-SUMO moiety, a highly purified SRSF1 with a molecular weight of around 28 kg/mol was obtained. The protein was analyzed by sizing chromatography and it was found that SRSF1 would form a polymer structure in the solution. According to Expasy bioinformatics analysis, SRSF1 is extremely unstable. Purification of full-length SRSF1 protein provides an opportunity to study mRNA splicing in vitro.
Key words: protein purification; serine/arginine-rich splicing factor 1(SRSF1); SUMO; SUMO protease; bioinformatics; chromatography
Introduction
Alternative splicing is tightly regulated in different tissues, cell types and differentiation stages, and dysregulation of this process has been associated with myelodysplastic syndrome, cancer, and autoimmune disease[1-3]. Spliceosome, which executes mRNA splicing, is a highly dynamical supramolecular complex. It consists of five small nuclear ribonucleoprotein particles (snRNPs, U1, U2, U4/U6, and U5) and more than 150 additional proteins[4]. Among them, serine/arginine-rich splicing factor family proteins are key regulators for spliceosome assembly and RNA binding. The three-dimensional(3D) structure of yeast spliceosome in different functional states has solved by cryo-electron microscopy in Yigong Shi’s research team, almost covering the whole splicing process[5].
Human serine/arginine-rich splicing factor 1 (SRSF1) is a 248aa protein containing two RNA recognition motifs (RRM1 and RRM2) that are responsible for its specific interaction with RNA, and a C-terminal serine/arginine-rich domain, which is involved in protein-protein interactions[6]. SRSF1 bound to short motifs (usually CN for RRM1 and GGA for RRM2), which were found in exonic regions, to activate or repress the inclusion of hundreds of exons[7-8]. As a prototype member of the serine/arginine family, SRSF1 is essential for cell survival, knocking out of which in human cells and rodents is lethal. Besides alternative splicing, SRSF1 also plays an important role in mRNA transcription, nuclear export, stability, and translation[9]. Recent works have solved the high-resolution 3D structures of RRM1/RRM2 binding with specific RNA motifs[7-8], but the structure of full-length SRSF1 remains to be determined.
Small ubiquitin-related modifier (SUMO) is a chaperon, with approximately 100 amino acids. When fused to N-terminal or C-terminal of target protein, it can dramatically enhance protein folding, solubility, and expression[10-11]. Thus, SUMO fusion technology has been widely used inEscherichiacoli(E.coli) expression of recombinant proteins. In the present work, we mainly want to find if the protein sequence of SRSF1 is highly conserved among different species, and if it is consistent with its pivotal role in cell survival. Bacterial expression system is established to purify and characterize full-length human SRSF1 in solution. Our work is necessary to get well diffracted protein crystals and study the function of SRSF1 in spliceosome construction and recognition of RNA motifsinvitro.
1 Materials and Methods
1.1 E.coli strains
The DH5α and BL21 (DE3) competent cells were prepared in the laboratory of Donghua University, China. All constructions were conducted inE.coliDH5α and BL21 (DE3) through transformation by using the heat shock method. The chemically competentE.colicells were prepared by using the Inoue method[12].
1.2 Construction of SRSF1 recombinant plasmid
The coding sequence of full-length human SRSF1 (NCBI reference sequence: NM_006924.5) was synthesized by Sangon Biotech (Shanghai, China) and optimized according to the codon usage preference ofE.coli. The optimized sequence was cloned into a modified pET-28a(+) vector (Novagen, USA) containing N-terminal His and SUMO tag (pET-28a-ppSUMO) through polymerase chain reaction and restriction sites for EcoRI and HindIII. The His-SUMO-SRSF1 recombinant plasmid was transformed into DH5α for amplification and verified by sequencing.
Primers used for polymerase chain reaction are
F: 5′-ATATAgaattcTCTGGTGGTGGTGTTATCCG-3′;
R: 5′-ATATAaagcttTTAGGTACGGCTACGGCTACG-3′.
1.3 His-SUMO-SRSF1 expression
The verified His-SUMO-SRSF1 recombinant plasmid was transformed intoE.coliBL21 (DE3) cells. After kanamycin resistance screening, a positive monoclone was picked into 4 mL lysogeny broth (LB) culture medium and incubated overnight on a shaking bed at 37 ℃ and 225 r/min. Then the culture medium was inoculated into 1 L LB liquid medium (Kana-resistant) and cultured at 37 ℃ and 225 r/min until the optical density at 600 nm (OD 600) reached 0.5. Then the isopropyl β-D-thiogalactoside (IPTG final concentration of 1 mmol/L) was added to induce recombinant protein expression at 37 ℃ and 225 r/min for 4 h.
1.4 Protein purification
Cells were harvested by centrifuge at 4 ℃ and 4 000 r/min and resuspended with precooled lysis buffer (20 mmol/L Tris, pH of 8.0, 400 mmol/L NaCl, and 1 mmol/L phenylmethanesulfonyl fluoride (PMSF)). Then the cells were crushed with a high-pressure cell crusher at 1 000 MPa for 3 times. After being centrifuged at 4 ℃ and 12 000 r/min for 1 h, the supernatant was collected. The NI-NTA column was balanced with a 20 times volume lysis buffer, and the supernatant was poured into the column slowly to fully combine the fusion protein with the resin. Then wash buffers (50 mmol/L Tris, pH of 8.0, and 400 mmol/L NaCl) containing different concentrations (10, 25, and 50 mmol/L) of imidazole were used to remove the miscellaneous proteins. Elute buffer (50 mmol/L Tris, pH of 8.0, 400 mmol/L NaCl, and 100 mmol/L imidazole) was used to elute the fusion protein, and strip buffer (50 mmol/L Tris, pH of 8.0, 400 mmol/L NaCl, and 250 mmol/L imidazole) was used to wash all the miscellaneous proteins from the medium and collect all the samples. The samples were collected separately and identified by 12% SDS-PAGE and western blot (HRP-conjugated 6×His antibody, HRP-66005, Proteintech) analyses.
An ultrafiltration tube with a molecular weight cut off of 10 kg/mol was selected to concentrate the eluents containing fusion protein. The concentrated solution was exchanged with imidazole free buffer (50 mmol/L Tris-HCl, pH of 8.0, 400 mmol/L NaCl, glycerol with a volume ratio of 5%, and 1 mmol/L dithiothreitol (DTT)) to remove imidazole. The fusion protein was centrifuged and concentrated at 4 ℃ and 4 000 r/min for enzyme digestion and purification. SUMO protease (6 × His tagged ULP1, 0.1 μg/mL) and 15 mL imidazole-free buffer were added into the concentrated His-SUMO-SRSF1 fusion protein and digested overnight at 4 ℃. After digestion, the sample was passed through the NI-NTA column twice. The solution was collected, and the digested products were determined by 12% SDS-PAGE.
An ultrafiltration tube with a molecular weight cut off of 3 kg/mol was selected to concentrate the digested protein to 250 μL at 4 ℃ and 4 000 r/min. A Hiload 16/60 Superdex200 column was selected to determine concentration and purity of the target protein. The column was first balanced with two buffer volumes (50 mmol/L Tris, pH of 8.0, 400 mmol/L NaCl, 1 mmol/L DTT, and 1 mmol/L ethylene diamine tetraacetic acid (EDTA)). Samples were loaded at a flow rate of 0.5 mL/min and a pressure of 0.3 MPa. The eluotropic samples at the absorption peak of UV280 were collected (all operations were carried out at 4 ℃) and detected by 12% SDS-PAGE. Finally, the protein samples were mixed, concentrated, and supplemented with glycerol at a volume ratio of 20% for storage at lower temperature.
1.5 Sequence alignment
Protein sequences of SRSF1 were downloaded from UniProt and aligned using Clustal Omega.
2 Results and Discussion
2.1 Clustal alignment of SRSF1 among species
SRSF1 contains two RNA recognition motifs and a C-terminal serine/arginine-rich (RS) domain as shown in Fig. 1(a). We aligned SRSF1 protein sequence among different species, and the results showed that it was really conserved, for the sequences of humans, mice, rats and several other species with a 100% identity (Fig. 1(b)). The high sequence conservation implies the irreplaceable roles of the protein.
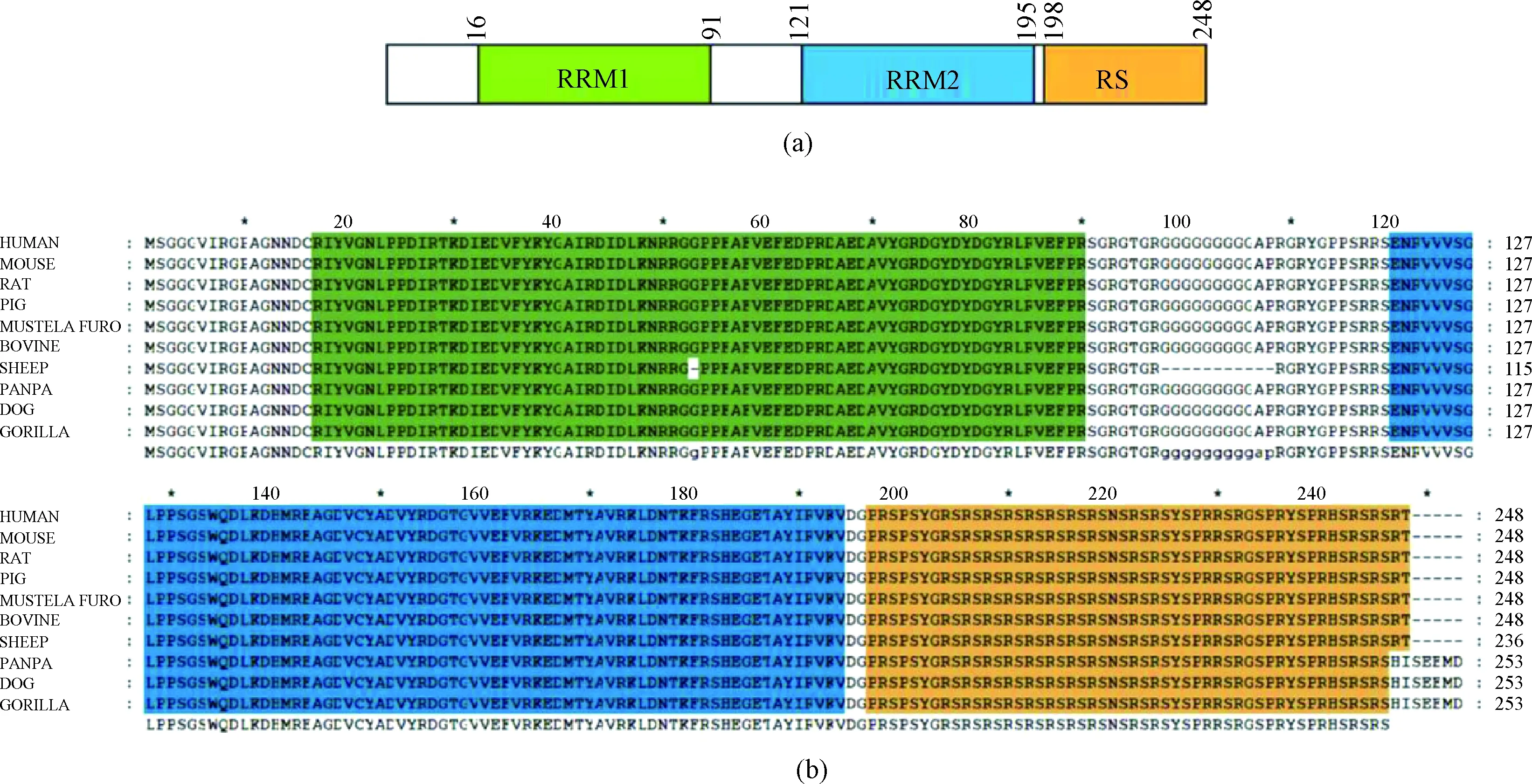
Fig. 1 Highly conserved SRSF1: (a) a schematic presentation of the functional domains of human SRSF1; (b) multiple sequence alignment of SRSF
2.2 Construction of His-SUMO-SRSF1 recombinant plasmid
In order to construct the recombinant plasmid, SRSF1 coding sequence was inserted into the pET-28a-ppSUMO vector (Fig. 2(a)). After double-enzyme digestion, the linearized vector was about 5 600 bp (Fig. 2(b)), and the target gene fragment was about 747 bp (Fig. 2(c)). All the target bands were indicated with an arrow.

Fig. 2 Construction of His-SUMO-SRSF1 plasmid: (a) map of His-SUMO-SRSF1; (b) pET-28a-ppSUMO vector digested by EcoRI and HindIII; (c) SRSF1 sequence digested by EcoRI and HindIII
2.3 Expression and purification of His-SUMO-SRSF1 fusion protein
His-SUMO-SRSF1 contains a 6×His-SUMO moiety and can be purified by Ni-NTA affinity chromatography. The measured molecular weight of His-SUMO-SRSF1 was about 46 kg/mol (Figs. 3 (a) and 3 (b)), consistent with the theoretical molecular weight in SDS-PAGE and western blot analysis. SDS-PAGE detection results are shown in Fig. 3(a). The fusion protein exhibited excellent solubility (lanes 3 and 4). It seems that there is almost no target protein in lane 5, indicating that the fusion protein was nearly all bound to the Ni-NTA resin. After being washed with imidazole buffer at the concentrations of 10, 25, and 50 mmol/L (lanes 6-8), most hybrid proteins were removed. Large amounts of His-SUMO-SRSF1 were eluted with imidazole at a concentration of 100 mmol/L (lanes 9-11). In the end, all target proteins were eluted with 250 mmol/L imidazole, and the protein purity was high as shown in Fig. 3(a). Qualitative detection of purified His-SUMO-SRSF1 was carried out by western blot analysis. The target band appears in the correct position about 46 kg/mol (Fig. 3(b)).
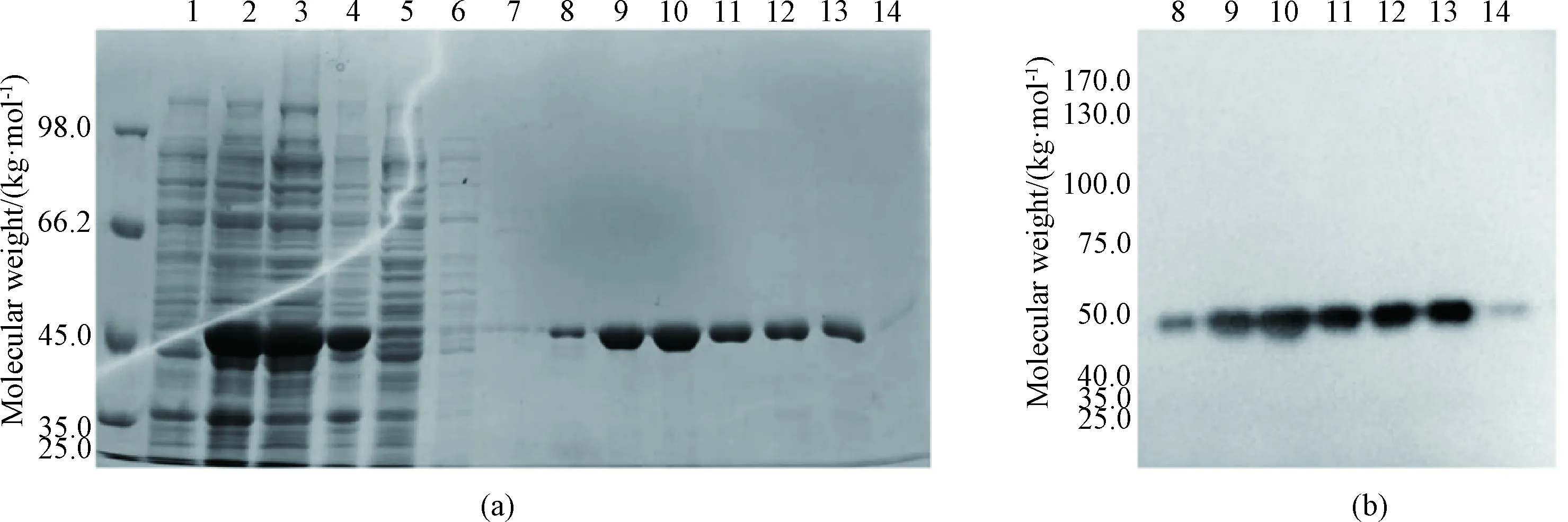
1—cell extract before IPTG induction; 2—cell extract after induction; 3—cell lysis supernatant; 4—precipitated protein; 5—flow through solution; 6—cell being washed with 10 mmol/L imidazole; 7—cell being washed with 25 mmol/L imidazole; 8—cell being washed with 50 mmol/L imidazole; 9-11—cell being eluted with 100 mmol/L imidazole; 12-14—cell being eluted with 250 mmol/L imidazole.
2.4 Cleavage of His-SUMO tag
The purified fusion protein contains a 6 × His-tag and an SUMO-tag. To obtain tag-free full-length SRSF1, it was incubated with SUMO protease overnight at 4 ℃ and loaded to NI-NTA affinity chromatography again to separate the target protein from SUMO protease and cleaved His-SUMO moiety. SDS-PAGE and western blot analysis were used to analyze the samples before and after digestion. The molecular weight of SRSF1 after digestion was about 28 kg/mol (Fig.4(a)), which was consistent with the theoretical value. Western blot analysis showed that almost all His-SUMO moieties were removed after digestion (Fig.4(b)), with an efficiency of 95%.

1—SRSF1 fusion protein before SUMO protease digestion; 2—SRSF1 after SUMO protease digestion; 3—SRSF1 passed through NI-NTA column.
2.5 Chromatography and bioinformatic analysis of SRSF1 protein
SRSF1 protein was further purified by Hiload 16/60 Superdex200 gel filtration chromatography column (GE, USA) in order to obtain high purity of the target protein. After separation, two distinct peaks exist as shown in Fig. 5(a) whereA280is the absorption wavelength with the highest absorption peak of proteins and phenols. The first peak (P1) appeared at 48 mL, which showed a molecular weight of 440 kg/mol. The second peak (P2) appeared at 88 mL, where showed a molecular weight of about 30 kg/mol, the same as the target protein. SRSF1 solution is heterogenous of both monomer and polymer. It was confirmed that P1 was SRSF1 polymer (Fig. 5(b)).
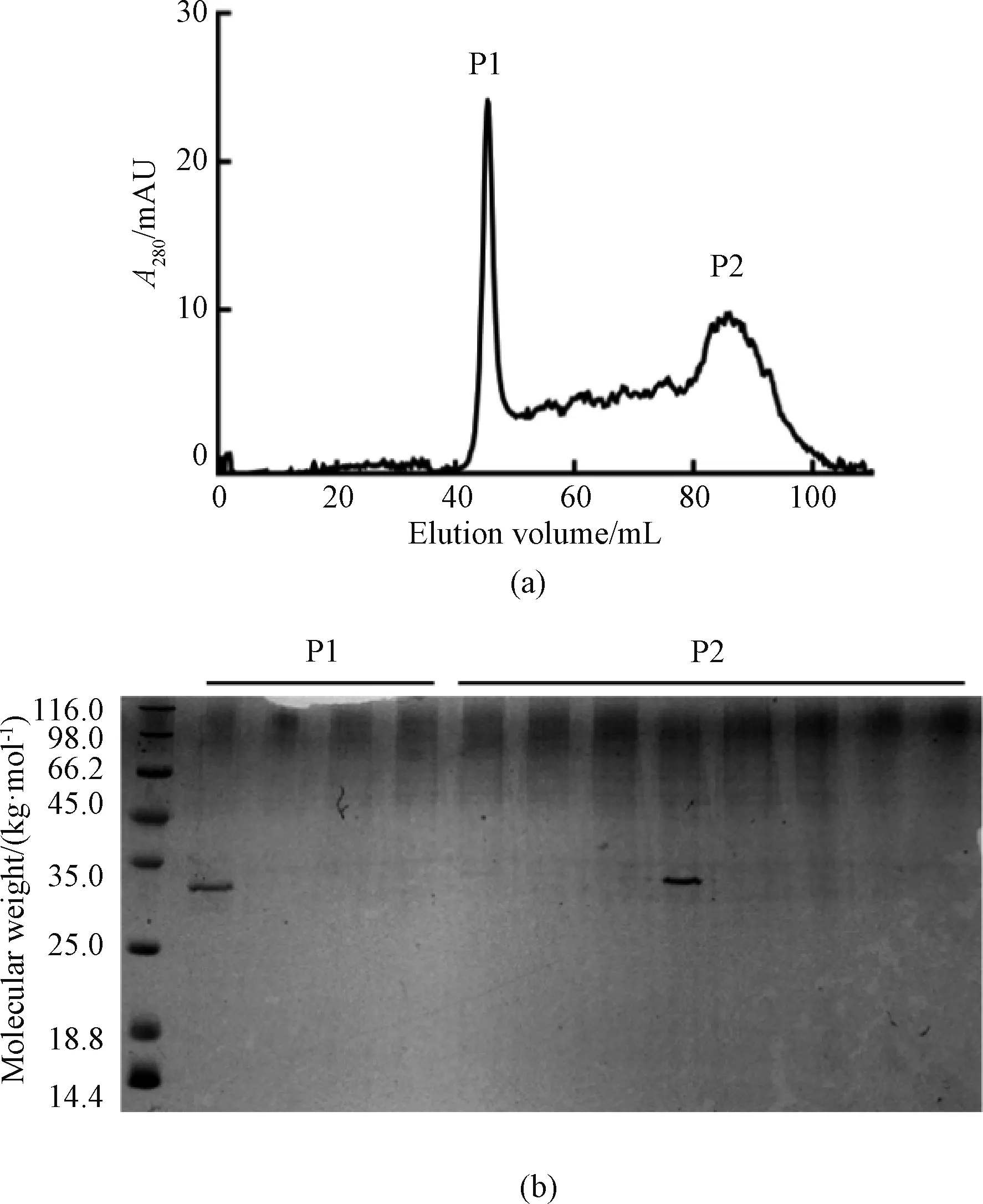
Fig. 5 Size exclusion of SRSF1: (a) Hiload 16/60 Superdex200 size exclusion; (b) 12% SDS-PAGE of SRSF1
Further analysis with bioinformatics software Expasy predicted that half time of SRSF1 was different in different species. It had the longest time in mammalianinvitro, and 20 h in yeast, and 10 h inE.colicells, respectivelyinvivo(Fig. 6(a)). This property provides an opportunity for the study of protein propertiesinvitro. What is more, the protein is extremely unstable in solution, and the instability index (II) is computed to be 79.73 (Fig. 6(b)).

Fig. 6 Physical properties prediction of SRSF1: (a) half-time prediction of SRSF1; (b) instability prediction of SRSF1
3 Conclusions
It was demonstrated that an SUMO moiety would help to enhance the expression, solubility and activity of recombinant proteins, and could be fully removed by SUMO protease to generate target proteins with the native structure. Due to its flexible inter-RRM linker (connecting region between RRM1 and RRM2), the 3D structure of full-length SRSF1 remains unclear. Here, an SUMO fusion system was established to purify full-length SRSF1 fromE.coli. After a series of expression and purification processes, large amounts of high-quality SRSF1 were obtained. The product can be used for protein crystallization andinvitrofunctional studies. This result will further reveal a new insight into the interactions between RRM1 and RRM2 in the full length, and also provide opportunity for RNA binding studies for the full length human SRSF1.
 Journal of Donghua University(English Edition)2022年5期
Journal of Donghua University(English Edition)2022年5期
- Journal of Donghua University(English Edition)的其它文章
- Preparation of Hydrophilic and Shrink-Proofing Wool Fabrics Through Thiol-Ene Click Chemistry Reaction
- Performance of Array-Type Noncontact End Gripper Based on Coanda Mechanism for Gripping Garment Fabrics
- Modeling and Analysis of Production Logistics Spatio-Temporal Graph Network Driven by Digital Twin
- Navigation Method Based on Improved Rapid Exploration Random Tree Star-Smart (RRT*-Smart) and Deep Reinforcement Learning
- Residual Network with Enhanced Positional Attention and Global Prior for Clothing Parsing
- Research Status and Development Direction of Smart Clothing Materials