
阿尔茨海默症发病机制及基于吡喃/啶酮骨架的多靶点药物的研究进展
2022-12-17 02:37刘雪艳综述查代君审校
福建医科大学学报 2022年5期
刘雪艳(综述),查代君(审校)
阿尔茨海默病(Alzheimer’s disease,AD)是一种与年龄相关的慢性疾病,也是导致痴呆的最常见的原因。作为一种神经退行性疾病,AD患者伴随认知和行为功能受损,并最终导致死亡。根据WHO的数据,目前全世界约有5 000万例AD患者[1]。预计到2050年,病例数将超过1.5亿,给社会造成重大负担[2]。中国AD患者的数量不仅位居世界第一,其增速也是全球最快的。据估计,我国60岁以上人群的AD发病率将从2010年的12%增至2050年的33%,治疗费用也将超过1万亿元[3]。随着我国老龄化进程的加剧,AD给患者的家庭带来沉重的经济、情感和精神压力,防治AD已经刻不容缓。
虽然AD的发病机制已研究了100多年,但确切的病因尚不清楚[4],普遍认为AD的发病与多种因素有关,如年龄、家族遗传和代谢性因素等。AD的主要病理特征如下:患者大脑皮质和海马区沉积的β-淀粉样蛋白(amyloidβ-protein, Aβ)形成大量老年斑(senile plaques, SP),神经元胞体内因Tau蛋白异常聚集出现神经原纤维缠结(neurofibrillary tangles, NFT),并伴有神经元的大量丢失[5-7]。此外,部分研究认为,神经炎症、氧化应激、生物金属的稳态失调和神经递质缺陷等与AD的发生和发展也密切相关[8]。多种病理因素的交互作用共同导致了记忆缺陷甚至是认知功能障碍,进而促进AD的进展[9]。
目前已发现许多影响AD发生发展的主要靶点,例如乙酰胆碱酯酶(acetylcholinesterase, AChE)、β-位点淀粉样前体蛋白切割酶-1(β-site amyloid precursor protein cleaving enzyme-1,BACE-1)、糖原合成激酶3β(glycogen synthase kinase-3β, GSK-3β)、单胺氧化酶(monoamine oxidase, MAO)、大脑中的生物金属离子、N-甲基-D-天冬氨酸(N-methyl-D-aspartate, NMDA)受体、5-羟色胺(5-hydroxytryptamine, 5-HT)受体、组胺受体的第三亚型(H3受体)和磷酸二酯酶(phosphodiesterase, PDE)[10-11]。这些靶点与各自的信号通路以及彼此之间发挥作用共同影响AD的进展,形成AD复杂而不明确的疾病网络(图1)。
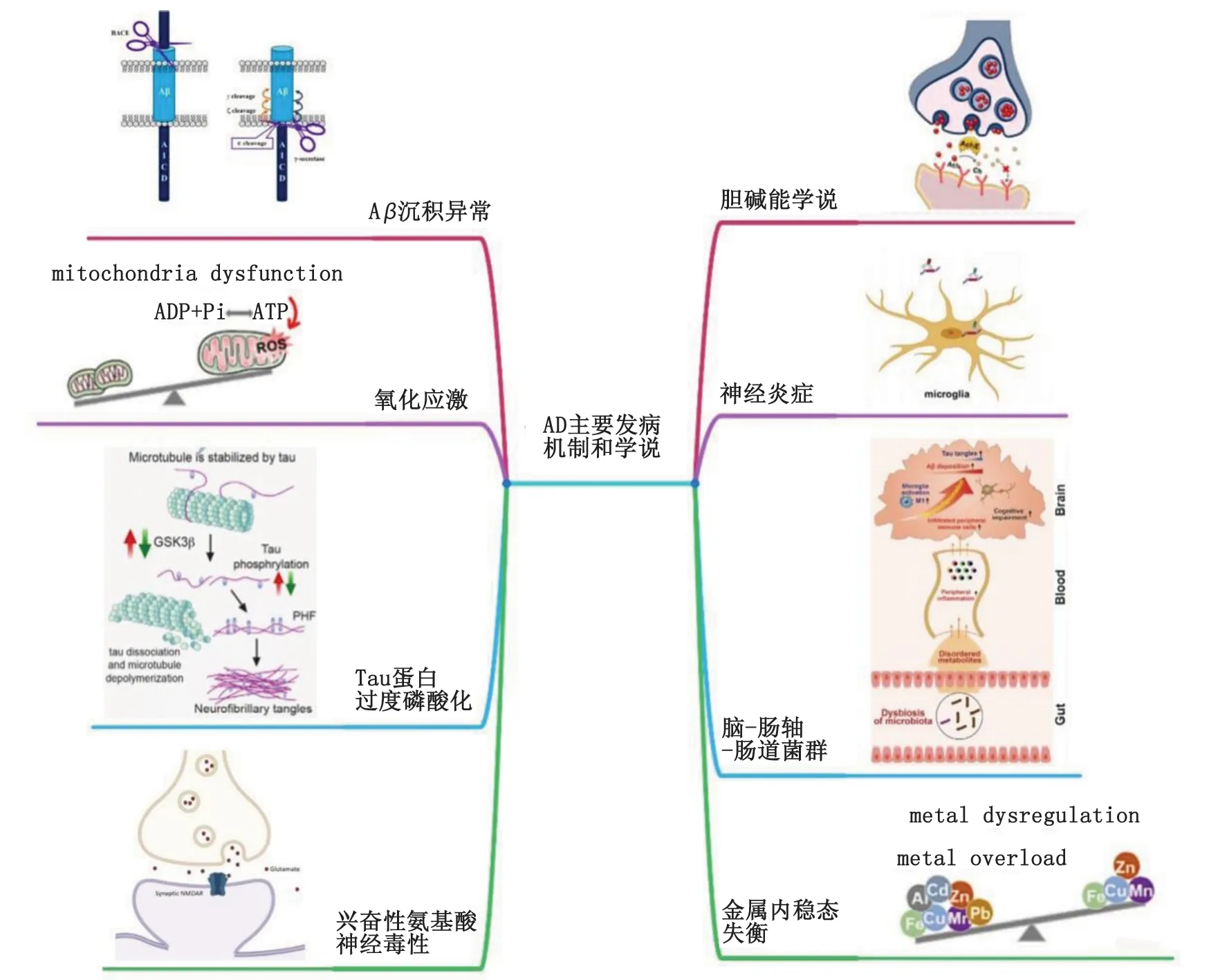
图1 阿尔茨海默病的主要发病学说及其靶点
1 发病机制
1.1 胆碱能假说 1976年由戴维斯和马洛尼首次提出胆碱能假说,这是目前AD研究中最为经典的假说。胆碱能假说将认知功能障碍归因于乙酰胆碱(acetylcholine,ACh)的消耗[12]。ACh作为一种参与学习和记忆的神经递质,在体内可被AChE和丁酰胆碱酯酶(butyrylcholinesterase, BuChE)水解而消耗。研究认为,低水平的ACh与AD患者的认知障碍密切相关,通过抑制AChE和BuChE来提高ACh水平是改善认知功能的有效策略[13]。健康成年人大脑中AChE的比例约为80%,其余为BuChE[14]。AChE有两个主要的结合位点,包括峡谷底部的催化阴离子位点(CAS)和峡谷顶部的外围阴离子位点(PAS)。大量证据表明,位于PAS的Trp279(eeAChE)或Trp286(huAChE)可加速Aβ斑块的形成并加重其神经毒性。BuChE主要定位于神经胶质细胞中,在人脑ACh的调节中也发挥着重要的作用。研究发现,在健康大脑中,大部分的ACh由AChE水解。但在受AD影响的大脑中,AChE水平显著降低,而BuChE活性增加,导致ACh主要由BuChE水解。不难看出,AChE和BuChE在AD的不同阶段所起的作用不同。在AD进展期,BuChE起主导作用,已成为治疗中重度AD的作用靶点。基于此,开发AChE和BuChE双重抑制剂有利于解决AD不同病理阶段的问题。此外,选择性BuChE抑制剂还可有效避免AChE抑制剂(acetylcholinesterase inhibitor, AChEI)的外周副作用,因此格外受关注[15]。
AChE是目前药物化学研究中防治AD的最主要的靶点和基本评价标准之一,抑制AChE可防止突触ACh的降解[16]。目前FDA批准的临床药物主要是AChEI,如多奈哌齐、加兰他敏和利凡斯的明。他克林作为第一个用于治疗AD的AChEI,于1993年批准上市,由于肝毒性已被停用,但其结构对新药的设计仍然具有指导意义。卫材公司和辉瑞公司研发的多奈哌齐是全球最畅销的AD药物,于1997年批准用于治疗轻中度AD,于2014年批准用于路易体痴呆的辅助治疗。根据目前的临床证据,多奈哌齐是一种安全且耐受性良好的药物,即使对严重的AD患者,也具有较好地改善认知功能的作用。目前,他克林和多奈哌齐仍被各研究小组深入研究,并常被用于体外酶活性试验和体内AD相关动物模型的药理实验阳性对照,以检测AChE的抑制效力和安全性。胆碱能假说和已批准的药物使AChEI在AD领域备受瞩目,针对AChE的单靶点策略和涉及AChE的多靶点策略成为AD潜在药物设计的共同思路。
1.2 Aβ异常沉积 Aβ假说是目前AD进展中最为流行的假说,由哈代和希金斯于1992年首次提出。Aβ级联反应学说认为:AD是由膜内淀粉样前体蛋白(amyloid precursor protein, APP)的异常水解和错误折叠所致的一种淀粉样变性疾病。APP是一种在大脑中高度表达的跨膜蛋白,其C端和N端分别通过神经元细胞的磷脂双分子膜层。一组分泌酶介导APP两种不同的代谢途径:α-分泌酶介导的非淀粉样蛋白途径导致片段容易降解,而β-分泌酶和γ-分泌酶介导的淀粉样蛋白途径导致Aβ肽的形成[17]。APP的降解主要通过α-分泌酶和γ-分泌酶催化的两步蛋白水解反应,不伴随物理损伤。而另有一小部分APP被β-分泌酶裂解,产生可溶性sAPPβ和CTFβ片段,该片段进一步裂解,产生APP胞内结构域AICD和Aβ(图2)[18-19]。Aβ由37~49个氨基酸残基组成,其中以Aβ40和Aβ42肽为代表。Aβ聚集增加、Aβ42/Aβ40比值增加、Aβ积累和Aβ清除减少等各种因素加速了大脑中SP的形成,进而导致AD的进展[20]。
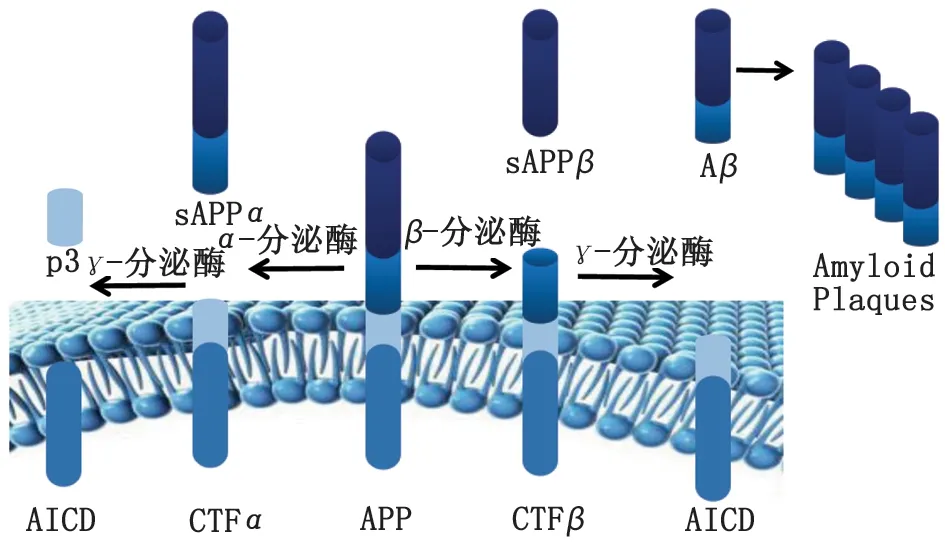
图2 Aβ的形成示意图
经典的Aβ假说表明,Aβ(特别是Aβ1-42)通过聚集成低聚物、原纤维,并最终形成斑块,进而引发线粒体功能障碍、慢性炎症和神经元损伤等一系列病理反应[21]。研究发现,Aβ可通过不同的机制诱导毒性作用。例如,Aβ在大脑中的积累会导致突触的丧失、神经元活动和突触传递的改变。此外,Aβ也参与了金属介导的氧化应激反应,还可以激活一系列病理事件,包括星形胶质细胞和小胶质细胞的激活,血脑屏障(blood brain barrier, BBB)的崩溃和微循环的变化等。
鉴于Aβ假说以及抑制Aβ沉积可干预AD进展的共识,BACE-1抑制剂治疗AD成为一个有吸引力的治疗策略[22]。然而,BACE-1抑制剂的研发现状却异常残酷。由于缺乏疗效和安全性不佳,百健和卫材公司Ⅲ期临床试验药物Elenbecestat于2019年终止研究;礼来公司Ⅱ期临床试验药物LY3202626由于缺乏具有统计学意义的疗效也于2021年宣布终止。
1.3 过度磷酸化的Tau蛋白 Tau蛋白假说是基于大脑细胞内的NFT,这是AD的另一个组织病理学标志[23]。Tau蛋白假说认为,Tau蛋白过度磷酸化在AD的发病过程中起关键作用。Tau蛋白是一种可溶性微管相关蛋白(microtubule-associated proteins,MAPs),其主要生理功能是与微管蛋白形成微管,维持微管稳定,促进微管聚集。当Tau蛋白被高度保守的苏氨酸-丝氨酸激酶GSK-3β过度磷酸化时,Ser396、Ser199和Ser413等位点的磷酸基从原始的2~3个增至5~9个,导致Tau蛋白无法与微管结合,造成微管的不稳定。同时,解离的Tau蛋白在神经元中积累和聚集,导致NFT的形成和神经元死亡[24]。自动高通量荧光成像系统研究表明,GSK-3β活性的异常增加还可导致β-分泌酶定位/缺陷和Aβ的分泌[25]。研究发现,GSK-3β在小胶质细胞和星形胶质细胞中均有表达,可促进细胞因子的产生,如肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)和白细胞介素1β(interleukin-1β, IL-1β)等,并可通过调节神经炎症过程促进神经疾病的发生和进展。此外,GSK-3β还可以一种独特的方式调节γ-分泌酶,诱导Aβ的形成,从而对神经元产生毒性。GSK-3β是上游Tau通路的主要特异性靶点,也是最受欢迎的小分子化合物研究靶点之一。抑制过度磷酸化的Tau蛋白聚集或阻断Tau蛋白的过度磷酸化是两种可能的策略。基于Tau蛋白聚集抑制剂对AD潜在的治疗作用使其越来越受到关注,目前已有多家制药公司正在对Tau蛋白抗体开展临床试验,如AC Immune与基因泰克公司联合开发的抗Tau蛋白单克隆抗体Semorinemab,强生公司针对靶向中区表位的致病性Tau单克隆抗体JNJ-63733657,以及Axon Neuroscience SE公司针对AD病理性Tau蛋白的活性肽疫苗AADvac1[26]。
1.4 神经炎症 神经炎症主要表现为中枢神经系统(central nervous system,CNS)中的胶质细胞的稳态失衡,是AD发病机制的关键因素。小胶质细胞作为CNS中的固有免疫细胞,在CNS的先天免疫反应中起着关键作用。在AD最初的病理过程中,小胶质细胞和星形胶质细胞发挥有益作用,如参与了淀粉样蛋白的清除。然而,随着疾病的进展,活化的小胶质细胞可表达促炎细胞因子(如IL-1β、IL-6 和TNF-α),伴随Aβ的清除能力下降,从而加剧神经退行性变。此外,在慢性神经炎症期间,BBB经常受到损害,进一步加重了神经炎症反应。同时,炎症环境可进一步促进SP和NFT的形成,导致AD的进一步恶化。因此,保持小胶质细胞的稳态可能是治疗神经炎症介导的神经退行性疾病的一种有效策略。新近研究发现,IL-3是星形胶质细胞-小胶质细胞串扰的关键中介物,可直接诱导神经元凋亡或放大局部炎症反应,可能是AD治疗的一个关键靶点[27]。集落刺激因子1受体(colony stimulating factor-1 receptor, CSF-1R)在CNS中能够直接参与小胶质细胞的分化及生长,并可减少蛋白沉积,因此靶向小胶质细胞的CSF-1R抑制剂在AD的治疗中极具潜能[28]。此外,主要表达在小胶质细胞上的髓系细胞触发受体2(triggering receptor expressed on myeloid cells-2, TREM2)不仅可影响Aβ和Tau蛋白的沉积,还可以参与炎症反应和代谢,也成为AD领域药物研发的潜力靶点[29]。然而,目前对于TREM2靶点的研究尚处于起步阶段,其中安进公司的VGL-101单克隆抗体和德纳利公司的DNL-919抗体处于临床前研究阶段,Alector公司研发的AL-002单克隆抗体正在进行临床Ⅱ期试验[30]。
1.5 氧化应激(oxidative stress, OS) 由于大脑对氧气的高需求和抗氧化防御系统的缺乏,使大脑非常容易受到OS的伤害。OS水平升高被认为是导致AD发病的早期事件之一,最终导致神经退行性变。大量证据表明,活性氧(reactive oxygen species, ROS)与线粒体氧化还原系统的失衡密切相关。从线粒体内膜泄漏的电子与氧原子之间发生反应产生高反应性的超氧阴离子,这些超氧自由基可以进一步进行自由基链反应,生成其他ROS,如过氧化氢和羟基离子[31-32]。研究发现,ROS的过度生成导致OS,OS在AD的发病机制和病理生理学中起关键作用。AD早期的OS可能是启动其他病理机制所必需的,如Aβ和Tau蛋白诱导的神经毒性。此外,异常浓度的生物金属离子(如Cu2+、Zn2+、Fe2+、Fe3+)共定位于Aβ斑块内,Aβ-Cu2+和Aβ-Fe2+/3+可以通过氧化还原循环诱导ROS的大量产生,进而加剧OS。基于ROS对蛋白沉积、生物金属离子代谢失调和线粒体功能障碍的影响,清除或阻止ROS的形成可能有助于阻止AD的进展。
人们普遍认为,OS与神经炎症是相互关联的。在ROS的刺激下,过度产生促炎细胞因子并诱导神经炎症。此外,慢性神经炎症会加重DNA损伤、突触功能障碍和Aβ负担,导致加速认知衰退和神经元死亡。因此,抗氧化剂和抗神经炎剂的应用可能为对抗AD提供互补作用。
1.6 金属离子稳态失衡 金属离子稳态失衡是AD患者大脑的一个重要特征。Cu2+、Fe2+、Zn2+和Fe3+是4种最重要的生物金属离子,它们的稳态失衡与AD的发病机制密切相关。Cu2+和Zn2+通过结合Aβ肽、影响Aβ聚集途径来诱导产生有毒的Aβ低聚物,而氧化还原活性金属Cu(Ⅰ/Ⅱ)和Fe(Ⅱ/Ⅲ)可通过Fenton反应引发OS并损伤神经元。因此,生物金属螯合剂可以下调高水平的生物金属,这可能是治疗AD的一种潜在的治疗策略。
1.7 MAO MAO是线粒体黄酮酶,由两种亚型(MAO-A和MAO-B)组成。MAO可催化各种生物胺和异生物胺的氧化脱氨反应,并在AD和帕金森病(Parkinson’s disease, PD)等神经退行性疾病中发挥重要作用[33]。MAO-A抑制剂临床上用于治疗焦虑和抑郁症,被认为是导致AD进展的危险因素,而MAO-B抑制剂用于治疗PD。MAO抑制剂可增加单胺神经传递,减少ROS形成和OS,从而发挥抗氧化、神经保护和认知改善等作用,对AD的治疗具有潜在的应用价值。
1.8 NMDA NMDA学说认为,作用于NMDA受体的谷氨酸刺激和谷氨酸相关兴奋性毒性的最佳平衡对于治疗AD至关重要。NMDA受体是一种离子型谷氨酸受体,由两个NR1亚基和两个NR2(NR2A-d)亚基或NR3亚基组成,当与兴奋性神经递质谷氨酸或调节剂甘氨酸结合时可被激活。NMDA受体在改变突触的可塑性、学习和记忆形成以及在将短期记忆巩固为长期记忆中起至关重要的作用[34]。而当NMDA受体被过量的谷氨酸过度刺激时,可导致谷氨酸相关的兴奋性毒性和细胞死亡。
研究认为,胆碱能和谷氨酸能神经元系统可相互影响,NMDA受体的过度激活与AD患者胆碱能神经元的退行性过程有关,例如,直接将NMDA 注入大鼠基底前脑导致神经元衰退和皮质中胆碱乙酰转移酶活性降低。因此,AChE和NMDA受体多靶点策略可以影响胆碱能和谷氨酸能系统,并协同拮抗神经退行性变。2015年批准由盐酸美金刚和盐酸多奈哌齐组成每日1次的固定剂量联合药物用于治疗AD[35]。因此,同时靶向AChE和NMDA受体,可能是个很有前途的策略。
1.9 5-HT 5-HT在情绪和抑郁中具有重要的生理功能,且与神经退行性疾病认知功能中的胆碱能系统有关[36]。5-HT被认为是一种抑制性神经递质。目前临床上选择性5-HT再摄取抑制剂(selective serotonin reuptake inhibitor, SSRI)以氟西汀和帕罗西汀为抗抑郁药物代表。该类药物通过抑制5-HT的神经元再摄取,增加突触间隙的5-HT浓度,从而改善抑郁症患者的情绪状态。根据其结构和功能特征进行分类,5-HT受体有7种亚型(5-HT1~5-HT7受体)。大脑中的5-HT1A、5-HT4、5-HT6和5-HT7受体与学习和记忆有关。5-HT1A受体在重度抑郁症的治疗中起重要作用,应用该受体的激动剂和拮抗剂都可能是AD的潜在治疗方法。如5-HT4受体参与记忆过程,该受体的部分激动剂可用于治疗AD的认知症状。5-HT6受体主要表达于大脑皮层和海马区,与学习和记忆过程相关,该受体的拮抗剂将有助于改善AD症状[37]。然而,目前靶向5-HT6抑制剂的研发却困难重重。如灵北制药的5-HT6受体拮抗剂Idalopirdine和Axovant公司的RVT-101,在临床Ⅲ期研究中心因未改善轻至中度AD患者的认知而终止临床实验[38]。
1.10 H3受体 组胺能系统由组胺及其受体组成,在维持CNS的稳态方面发挥重要作用,并参与平滑肌收缩、毛细血管扩张、胃酸分泌和外周炎症。组胺是一种分布在全身的内源性生物胺,在中枢作为神经递质,在周围作为局部介质,在肺、皮肤和胃肠道中浓度较高,并通过组胺受体发挥其生理功能。组胺受体是具有不同生物功能的G蛋白偶联受体,有4种亚型,即H1、H2、H3和H4受体[39]。H3受体可负向调节组胺和一些与认知相关的神经递质的释放,如ACh、去甲肾上腺素、多巴胺和血清素。H3受体拮抗剂可增加大脑中ACh和其他神经递质的含量,有利于对抗AD等神经退行性疾病[40]。
1.11 PDE 细胞内的第二信使环磷酸腺苷(cyclic adenosine monophosphate, cAMP)和环磷酸鸟苷(cyclic guanosine monophosphate, cGMP)对于细胞活动起着重要的调节作用。PDE可水解cAMP和cGMP,从而终结其传导所产生的生化作用。PDE有11个同工酶家族,从PDE1到PDE11,其中大多数有几个亚型。根据目前的估计,人类总共有100个特定的PDE。在这些家族中,PDE4、PDE7和PDE8是cAMP水解的特异性酶,而PDE5、PDE6和PDE9是cGMP水解的特异性酶。PDE1、PDE2、PDE3、PDE10和PDE11是一种双底物酶,可以同时水解cAMP和cGMP。具有降解第二信使能力的PDE,是神经可塑性和神经保护中信号转导的重要调节因子。因此,具有上调cAMP和cGMP浓度能力的PDE抑制剂作为治疗AD认知能力下降的潜在药物越来越受关注[41]。长春西汀是一种用于治疗痴呆和认知障碍的PDE1抑制剂,于1980年获得批准,但据报道它对AD患者的认知障碍无效。大冢制药公司的西洛他唑是一种口服PDE抑制剂,于1988年推出,用于改善各种慢性动脉阻塞症状,与多奈哌齐联合治疗轻中度AD。来自美国国家精神健康研究所的罗利普兰是一种PDE4D抑制剂,Ⅱ期临床试验证实其在记忆巩固中发挥了关键作用,用于治疗重度抑郁症。PDE5作为一种特殊的cGMP水解酶,只有一种亚型,即PDE5A,分布于大脑的海马、皮质和小脑。PDE作为新的治疗靶点,引起了众多学者的广泛关注,成为一个新的研究热点,选择性PDE4和PDE5抑制剂因分布在海马而受到格外的重视[42]。
1.12 脑—肠轴—肠道菌群假说 肠道微生物群是人体肠道中的细菌、病毒和真菌共生系统,影响人体消化、肠道生物合成、新陈代谢和炎症等生理过程。肠上皮屏障(intestinal epithelial barrier,IEB)是由单层上皮细胞构成黏膜界面,其功能是限制病原体和抗原的进入,为宿主提供生理和防御支持。肠血管屏障(gut-vascular barrier,GVB)由上皮细胞下方的细胞构成[43],为第二道防线,起到控制抗原易位并阻止肠道微生物群进入的功能。肠道菌群失调可诱导IEB通透性改变,导致微生物代谢物的渗漏和外周炎症,其产生的炎症因子可穿过BBB激活小胶质细胞的过表达,导致神经炎症和AD的进展。我国新近批准的一类新药甘露特钠胶囊(GV-971)通过重塑肠道菌群,降低异常代谢产物,阻止外周炎性向大脑浸润,从而抑制神经炎症和改善认知障碍[44]。
AD因具有复杂的发病机制和病理生理过程,导致其治疗困难,并给患者及其家属带来沉重的负担。因此,迫切需要研发有效的抗AD药物。世界各地的研究小组为克服AD做出了巨大的努力,发现了许多与AD相关的靶点,并试图解决这一科学难题。本综述讨论了12个与AD相关的机制学说,其中胆碱酯酶、BACE-1、GSK-3β、TREM2和CSF1R等成为研究的热门靶点,载脂蛋白E(apolipoprotein E, APOE)因与基因携带相关,又与Aβ沉积、Tau蛋白沉积、神经炎症以及线粒体功能和代谢等高度关联,也颇具潜能[45]。新近研究发现,溶酶体的“酸化”障碍所致的神经元细胞死亡极可能是AD进展的全新机制[46]。随着对AD疾病进展和分子机制的深入研究,开发通过多种作用机制抗AD的药物值得期待。
2 多靶点导向的药物研发策略
目前临床上传统的AD药物主要分为胆碱酯酶抑制剂(多奈哌齐、利凡斯的明、加兰他敏和石杉碱甲)和非竞争性NMDA拮抗剂(美金刚)。此外,2014年12月,食品药品监督管理局(Food and Drug Administration,FDA)批准了治疗AD的组合药——Namzaric,该药结合了盐酸美金刚胺缓释剂和盐酸多奈哌齐,用于治疗中重度AD。2019年11月,国家药品监督管理局(National Medical Products Administration, NMPA)有条件批准一类新药甘露特钠胶囊(GV-971)上市,用于临床治疗轻中度AD。2021年6月,FDA限制性批准靶向Aβ的单克隆抗体阿杜那单抗治疗AD。然而,现有上市的药物虽有一定程度的治疗作用,但并不能阻止或逆转疾病的进展,且存在一定的不良反应[47](表1)。

表1 目前抗AD上市药物的作用靶点及局限性
AD是一种发病机制复杂、病理表现多样的神经退行性疾病,但传统的AD治疗策略长期关注于“一个分子,一个靶点”的模式,很难改变AD的进展。近年来,多靶点定向配体(multi-target-directed ligand, MTDL)在AD治疗中显示出巨大的应用前景。
多靶点配体是将两个或多个关键药效团片段结合在一个分子里,这个分子与多个靶点均具有较好的亲和力并产生多靶点活性[48]。因此,MDTL策略为病因复杂的神经退行性疾病开辟了新的治疗途径。目前有相当数量的多靶点抗AD的小分子化合物被研发出来,有望成为改善疾病的治疗药物。胆碱酯酶学说作为经典的AD学说,对AD药物的研发具有积极的参考意义。世界各研究小组通过化学合成手段,设计合成一系列在靶向胆碱酯酶基础上具有多靶点抗AD的类药分子。
2018年,JIANG等[49]设计并合成了一类新的GSK-3β/AChE双靶点抑制剂。其中,化合物1(图3)在hAChE(IC50为6.5 nmol/L)和hGSK-3β激酶活性(IC50为66 nmol/L)方面显示出纳摩尔抑制作用。在20 μmol/L下,化合物1对Aβ自聚集显示出良好的抑制作用(抑制率为46%)。蛋白质免疫印迹(Western-blot, WB)分析显示,化合物1抑制小鼠神经母细胞瘤N2a Tau细胞中Tau蛋白的过度磷酸化。体内研究证实,化合物1能显著改善东莨菪碱导致的ICR小鼠的认知障碍。SHARMA等[50]通过拼接多奈哌齐中的N-苄基哌啶药效团,设计合成一系列5-苯基-1,3,4-口恶二唑多靶点衍生物,该类衍生物对hAChE、hBChE和β-分泌酶-1(hBACE-1)表现出较强的抑制作用,特别是化合物2(图3)对所有靶标均表现出平衡的抑制特性。SANG等[51]通过拼接利凡斯的明和MAO抑制剂的药效团,合成一系列新型邻氨基甲酰亚铁酰胺衍生物。体外生物学评价表明,化合物3(图3)具有hBChE抑制(IC50为0.97 μmol/L)和选择性MAO-B抑制作用(IC50为5.3 μmol/L),对Aβ诱导的自聚和解聚具有较好的抑制作用,抑制率分别为58.2%和43.3%。体内研究表明,化合物3对三氯化铝介导的斑马鱼运动障碍具有恢复作用,并对Aβ1-40引起的血管损伤具有显著的保护作用。此外,化合物3可改善东莨胆碱诱导的认知障碍。LIU等[52]通过对多奈哌齐的药效基团和天然来源的去氢骆驼蓬碱进行分子杂交,合成了一系列新型衍生物。研究发现,所有衍生物均具有显著的抗AChE活性,且对BuChE具有良好的选择性。化合物4(图3)具有强大的抗AChE活性(IC50为0.27 μmol/L)、选择性BuChE抑制活性(IC50为20.82 μmol/L),以及GSK-3β抑制活性(IC50为6.78 μmol/L)。分子对接研究和分子动力学模拟结果表明,化合物4可以与AChE和GSK-3β形成稳定的相互作用。在Tau(P301L)293T细胞模型中,化合物4可有效降低Tau蛋白Ser396位点的过度磷酸化。此外,化合物4对SH-SY5Y、HEK-293T、HL-7702和HepG2细胞系表现出极低的细胞毒性。
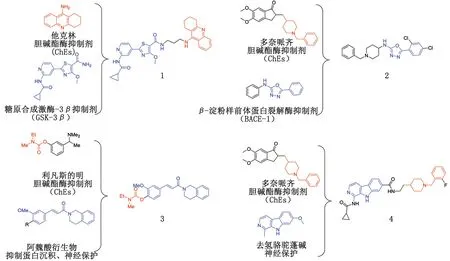
图3 基于多靶点策略设计的类药分子
AD错综复杂的发病机制使多靶点治疗药物的研发不限于胆碱酯酶。表2汇总了部分不依赖胆碱酯酶、已进入临床Ⅱ/Ⅲ期的多靶点治疗AD的代表药物。

表2 小分子抗AD药物临床研究进展
3 吡喃/啶酮骨架抗AD研究进展
杂环作为生命科学和生物化学之间的桥梁,为探索药用分子提供了不同的化学空间,受到全球范围内的极大关注。在这些杂环中,化合物5(羟基吡喃酮)及其电子等排体化合物6(羟基吡啶酮)是药物设计和发现的重要骨架。例如化合物7(去铁酮7),是一种口服活性的3-羟基吡啶-4-酮衍生物,多年来临床上一直用于铁超载地中海贫血患者的铁螯合剂[53](图4)。

图4 羟基吡喃酮、羟基吡啶酮和去铁酮的化学结构
羟基吡喃酮/吡啶酮作为一类重要的含氧杂环核,具有广泛的药理活性,如抗癌、抗炎、抗氧化、金属螯合、抗菌和抗真菌作用等,特别是其具有的抗氧化、抗炎和金属螯合作用可有效对抗AD的发病进程。同时,羟基吡喃酮/吡啶酮骨架具有分子量小和可修饰位点多的特点,除了3位羟基和4位羰基是螯合金属的必须基团外,其他环上2、5、6位都可以进行结构修饰,特别是1位的氧可以置换成氮、硫等杂原子,进一步促进其通过BBB[54](图5)。此外,通过其他多样性基团的引入,可以有效地改变其生物利用度、化合物的理化性质和生物特性,并可以通过拼接、融合等其他抗AD的活性基团,设计合成具有多种抗AD作用的类药分子,因此,羟基吡喃/吡啶酮在多靶点抗AD中的药物设计中具有极大的发展潜能。
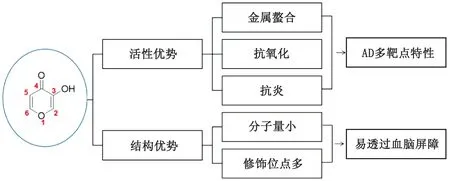
图5 吡喃酮骨架的活性与结构优势
白藜芦醇是一种天然存在的二苯乙烯类化合物,由于其具有抑制Aβ聚集和清除自由基等多种功能,目前已作为抗AD药物进入临床试验。CHENG等[55]将3-羟基吡喃-4-酮的结构融入白藜芦醇中,得到一系列白藜芦醇杂交体作为新型多靶点化合物。体外生物学评价表明,这些化合物具有抑制自诱导的Aβ1-42聚集、抗氧化和金属螯合活性。其中,化合物8(图6)对ABTS+自由基的清除活性强于维生素E类似物Trolox,对Aβ1-42聚集呈现微摩尔级别的抑制[IC50为(7.20±0.72)μmol/L],优于阳性对照物白藜芦醇[IC50为(11.89±2.52)μmol/L]。羟基吡喃酮基团的引入,赋予化合物8良好的金属螯合活性,可有效对抗Fe3+和Cu2+诱导的Aβ聚集。HU等[40]采用药效团结合策略,设计并合成一系列新型的2-苯乙烯基-5-羟基-4-吡喃酮衍生物,作为MTDL来治疗AD。化合物9(图6)在保留苯氧烷基胺骨架具备的H3受体拮抗作用(IC50为8.25 nmol/L)的同时,也呈现出羟基吡喃酮骨架所具备的优秀的金属离子螯合能力,比Trolox更强大的ABTS+清除能力以及高效Aβ自聚和Cu2+诱导的聚集抑制作用以及对Aβ自聚/Cu2+诱导聚集的分解能力。DGACHI等[56]基于曲酸和他克林的药效团设计合成一系列衍生物,其中化合物10(图6),得益于羟基吡喃酮骨架的贡献,具有强大的抗氧化能力(TE为4.79),并对Aβ1-40诱导的SH-SY5Y具有显著的神经保护作用。同时,经过结构修饰,化合物10的肝毒性低于他克林,并呈现出中等的AChEI[IC50为(4.52±0.24)μmol/L]。XU等[57]采用了药效团组合策略,设计合成了一系列去铁酮-白藜芦醇杂交体,评价其多功能抗AD的作用。体外生物活性评价表明,大部分化合物对Aβ1-42自诱导聚集具有较好的抑制作用,且具有较强的金属螯合能力和抗氧化活性。化合物11(图6)继承了羟基吡啶酮具有的ABTS+清除活性和金属螯合作用,还呈现出优于白藜芦醇的抑制Aβ1-42聚集[IC50为(10.72±0.5)μmol/L]作用。对接研究表明,化合物11可以与Aβ1-42蛋白的C端形成氢键发生相互作用。
香豆素作为MAO-B抑制剂的优势骨架,目前已被广泛研究。ZHANG等[58]设计合成一系列羟基吡啶酮和香豆素的衍生物,表现出优异的铁离子螯合作用和中等至良好的抗MAO-B活性。化合物12(图6)对MAO-B表现出较好的抑制作用(IC50为14.7 nmol/L),优于阳性对照药帕吉林(IC50为85.8 nmol/L)。同时,对U251细胞具有良好的保护作用,可显著改善东莨菪碱诱导的AD小鼠的认知功能障碍。与之类似,JIANG等[59]通过将羟基吡啶酮与香豆素结合,设计了新的杂交种,所有化合物均继承了母体化合物所具备的MAO-B的抑制活性和铁螯合活性。化合物13(图6)对MAO-B抑制作用的IC50为87.9 nmol/L,对Aβ1-42诱导的PC12细胞损伤具有细胞保护作用。Morris水迷宫测试显示,化合物13显著改善了小鼠的认知障碍。MI等[60]以香豆素和羟基吡啶酮为先导化合物,通过整合关键药效团设计合成了一系列双靶点杂交体,其中化合物14(图6)在保留羟基吡啶酮良好的铁离子螯合活性的基础上,具有优秀的MAO-B抑制活性(IC50为0.68 μmol/L)。BORTOLAMI等[61]设计了一系列去铁酮衍生物作为AD的潜在多功能化合物15(图6),其将去铁酮结构和2-氨基吡啶、2-氨基嘧啶或2,4-二氨基嘧啶等具有胆碱酯酶抑制活性基团用长烷烃链连接,以获得具有抑制胆碱酯酶、抑制Aβ聚集、螯合金属等多种活性。此外,该类化合物对于人脑星形胶质母细胞瘤细胞(U-87MG)具有较低的毒性。SHENG等[62]通过将3-羟基-4-吡啶酮和H3受体拮抗剂进行杂交,设计合成了一系列新型的1-苯基-3-羟基-4-吡啶衍生物。化合物16(图6)具有优于阳性对照物姜黄素的纳摩尔级别的H3受体拮抗作用[IC50为(2.85±0.22)nmol/L],并能有效阻断Aβ1-42原纤维的形成,具有良好的铜和铁螯合性能,以及较好的自由基离子清除活性。此外,体内试验研究表明,化合物16在0.5 h内脑内浓度达到207 ng/mL,脑/血浆暴露比为0.69,说明该化合物能够过BBB,这对于CNS的药物来说,极其关键。
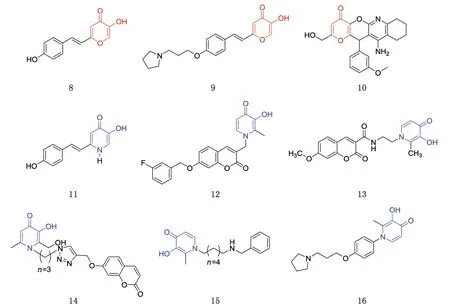
图6 基于吡喃酮及类似结构的多靶点抗AD潜力化合物
综上所述,通过合理的引入吡喃/啶酮及类似结构,可成为多功能抗AD小分子的优选结构。通过设计合成全新的吡喃/啶酮化合物,并对其进行活性评价和构效关系研究,为含有吡喃/啶酮母核的衍生物开发成具有抗AD的类药分子提供了参考依据。
在过去的10 a,多靶点药物发现策略在神经退行性疾病、癌症或心血管疾病等复杂疾病的药物研发中具有很大的潜力。AD的一个独特而复杂的特征就是不同信号通路中的相关靶点可发生相互作用,并形成一个疾病网络。考虑到AD的复杂的发病机制和单靶点药物的缺点,多靶点药物相对于单靶点药物,能够有效协同多个靶标,从而更好地影响疾病网络和控制AD进展。因此,多靶点设计策略是当前AD治疗研究的一个重要方向。然而,MTDL的设计也给药物化学家在优化活性和理化性质方面提出了一些挑战,特别是MTDL设计的化合物要求对每个靶标都有较好的作用,而在许多情况下,达到相似的效力(即调控两个靶点在相同浓度范围内)是一项具有挑战性的任务。此外,由于MTDL在大多数情况下是通过整合两个或更多选择性配体的结构元素而设计的,分子量比大多数商业药物更大、亲脂性也更强,因此可能导致口服吸收不良。另一方面,在AD等CNS疾病相关的药物发现过程中,另一个挑战是克服BBB。分子量(MW)、亲脂性(logP)、极性表面积(PSA)以及与P-糖蛋白外排转运体(P-gp)的相互作用都会影响分子对BBB的渗透能力。一般认为,分子量<450、PSA<90 Å2的药物具有较好的血脑通透性[63],而羟基吡喃酮/吡啶酮具有分子量小、PSA小以及结构易修饰的优点,使其在神经退行性疾病的药物设计中值得探索。随着计算机辅助药物设计在新药研发中的助力,可进一步发掘以羟基吡喃酮/吡啶酮为骨架的疗效更好、生物利用度更佳的多靶点抗AD的候选药物分子,以加快AD药物的研发。
猜你喜欢
化学工程师(2022年5期)2022-05-11
中老年保健(2021年3期)2021-12-03
中国生殖健康(2020年7期)2020-12-10
中成药(2018年2期)2018-05-09
新乡学院学报(2016年6期)2016-12-01
当代化工研究(2016年9期)2016-03-20
合成化学(2015年9期)2016-01-17
烟草科技(2015年8期)2015-12-20
医学研究杂志(2015年7期)2015-06-22
同位素(2014年2期)2014-04-16
