
伴肾小球薄基膜改变的IgA肾病非胶原Ⅳ基因变异分析
2022-12-19 01:46余学问梁莹莹曾又佳徐洲稳邵牧民
临床与实验病理学杂志 2022年10期
徐 华,余学问,梁莹莹,李 芸,曾又佳,张 露,徐洲稳,李 霞,邵牧民
IgA肾病在原发性肾小球疾病中占25%~50%。Qazi等[1]报道约19%的薄基膜病伴IgA肾病,2018年国际胶原Ⅳ相关肾病建议性新分类指出既往认为薄基膜病预后良好的观点需改变,也有文献报道合并高血压或肥胖等临床因素的薄基膜病预后不良[2-3]。近年,IgA肾病中肾小球薄基膜情况逐渐得以重视。约40%薄基膜病存在胶原Ⅳ基因变异[4-7],本组前期实验发现约6%的IgA肾病伴薄基膜病,部分具有胶原Ⅳ基因变异,非胶原Ⅳ基因变异情况尚不清楚。本实验利用全外显子组及Sanger测序技术筛选伴薄基膜改变的IgA肾病的变异基因,并对候选变异基因进行致病性评估,首次探索该类疾病中非胶原Ⅳ基因的变异及临床病理意义。
1 材料与方法
1.1 一般资料收集深圳市中医院病理科诊断的6例伴薄基膜改变的IgA肾病,患者年龄34~52岁,中位年龄41岁。根据临床表现、病理学形态、免疫荧光及透射电镜检查,按照本电镜室建立的正常成人肾小球基膜厚度标准[(340±70) nm]直接测量基膜厚度,结合文献关于薄基膜病变评估标准[8-9],本实验定义基膜厚度小于270 nm为薄基膜改变。收集患者临床信息包括:年龄、性别、家族史,做肾脏病理穿刺活检及近期随访时的血尿、蛋白尿、肾功能指标、血压,肾外表现如听力、眼、神经发育及运动功能给予相应专科检查,肾脏影像学检查确定有无囊肿、畸形或发育异常,排除其它系统性疾病。本实验经我院医学伦理委员会审查及批准(批件号K2019-027-01),患者均知情同意。
1.2 免疫荧光和免疫组化冷冻组织5 μm厚切片,以FITC羊抗人荧光抗体行直接免疫荧光染色,检测肾组织中IgG、IgA、IgM、C3、C1q、Fibrinogen、Kappa、Lambda(Dako公司)沉积部位、强度和分布特点。FN免疫组化染色采用EnVision法。穿刺组织2 μm厚切片脱蜡至水,EDTA热修复,3%H2O2阻断内源性过氧化物酶,5%BSA室温封闭,染色步骤采用Leica bond-Ⅲ全自动免疫组化仪按操作说明书进行,一抗为Fibronectin(RAB-0071,福州迈新公司),二抗为Leica原装,DAB染色,苏木精复染,脱水透明,封片观察。
1.3 全外显子组测序及变异分析提取外周血单个核细胞总DNA(QIAamp DNA Blood Mini Kit,QIAGEN公司,德国),制备文库,对外显子及临近剪切区捕获和富集,使用BGI-MGISEQ-2000高通量测序平台进行检测。总体覆盖度为95%,目标区平均测序深度>110X。测序片段通过BWA与UCSC hg19人类参考基因组比对,使用GATK(GenomeAnalysis Tool kit)行碱基质量值校正SNV及INDEL。对照变异数据库(dbSNP,1000 genome,ESP6500,GnomAD等)得到过滤后的突变频率,仅列出频率小于1%的变异。ClinVar(www.ncbi.nlm.nih.gov/clinvar)、OMIM(www.omim.org)、HGMD(www.hgmd.org)疾病数据库检索,采用Phylop(compgen.bscb.cornell.edu/phast)同源序列多重比对物种间保守性。根据检索文献及数据库,重点筛选肾小球基膜、足细胞及蛋白尿相关基因的变异。利用SIFT(sift.jcvi.org)、Polyphen2(genetics.bwh.harvard.edu/pph2)、MutationTaster(www.mutationtaster.org)、CADD(cadd.gs.washington.edu)、Varsome(varsome.com)功能学分析平台,参照美国医学遗传学及基因组学学会(The American College of Medical Genetics and Genomics, ACMG)标准,进行变异的致病性注释。按照人类基因组组织基因命名委员会(HGNC)及人类基因组变异学会(HGVS)规范命名。
1.4 Sanger测序验证FN1引物序列:上游5′-CAGTGAGGTAGGCAGGGAAG-3′,下游5′-TTT TAGCCAGACCCCAGATG-3′;上游5′-GGCTCCGTT GAATGGACTT-3′,下游5′-CCAAATAAAATGTT GAGGGA-3′;NUP160引物序列:上游5′-AGACTGA CAACCCATACA-3′,下游5′-GTTCCCGCAGAT AGAGT-3′;ITGA3引物序列:上游5′-TCTC CCCGCTCCAGTTC-3′,下游5′-TGGGTCCATTTGT TATCC-3′。扩增条件:96 ℃预变性5 min,96 ℃变性20 s,52 ℃退火30 s,72 ℃延伸30 s,合计35个循环,72 ℃延伸10 min。扩增产物经分离纯化后使用ABI3730xl测序仪测序,使用Phred/phrap软件进行SNP分析,使用SeqMan软件进行序列比对,使用Chromas软件查看峰图。
1.5 氨基酸链空间结构及蛋白结构域分析采用SwissModel(www.swissmodel.expasy.org)平台分析FN1突变位点对应的氨基酸链空间结构位置,用IBS.Biocuckoo(ibs.biocuckll.org/)软件构建FN1蛋白结构域图,采用String蛋白互作网络数据库(string-db.org/)分析FN1、NUP160、ITGA3蛋白之间的交互作用。
2 结果
2.1 临床特征例1女性,37岁,以间断肉眼血尿入院。隐血3+,尿蛋白2+,尿蛋白1 412 mg/24 h,肌酐87.8 μmol/L,ANA谱、ANCA、免疫、肝功能、电解质、凝血功能、血糖、血脂、风湿、血沉、抗磷脂酶A2受体抗体、β2微球蛋白、血免疫固定电泳、自身免疫性血管炎抗体检测均阴性;伴皮肤斑丘疹,未见听力、眼等肾外异常;未发现囊肿、畸形或泌尿生殖系统发育异常;骨骼系统及神经发育未见异常;否认家族肾病史。后经ACEI类药物及中药方剂治疗后,随访时尿蛋白减少,最低491.4 mg/24 h,血肌酐83 μmol/L。例2女性,34岁,以体检尿检异常入院。隐血+,尿蛋白波动1+~2+,尿蛋白531 mg/24 h,肌酐72 μmol/L,体液免疫IgA 4.26 g/L、IgG 16.71 g/L、IgM 2.40 g/L,N-乙酰氨基葡萄糖苷酶11.7 U/L,尿量2.6 L/24 h,尿素150.8 mmol/24 h,尿钠72.0 mmol/24 h,尿氯71.5 mmol/24 h,无机磷7.80 mmol/24 h;抗核抗体(±)、核颗粒型1 ∶100(+),其余临床实验室检查均阴性;伴皮肤斑丘疹,未见听力、眼等肾外异常;影像学检查、骨骼系统及神经发育未见异常;其兄有慢性肾衰竭史,否认父母有肾病史。经ACEI类药物及中药方剂治疗后,随访时尿蛋白波动,最低286 mg/24 h,血肌酐78 μmol/L。
2.2 病理检查例1:肾脏穿刺组织19个肾小球,其中2个球性硬化,4个节段性硬化,2个球囊黏连,未见新月体或坏死,轻度系膜增生(系膜基质增多及系膜细胞增生)。肾小管轻度萎缩,间质纤维化及单个核细胞浸润占肾皮质5%~10%。肾小球小动脉未见明显异常,个别小叶间动脉轻度内膜纤维化。免疫荧光:IgA及C3系膜区(3+),IgG、IgM、C1q、Fibrinogen、Kappa、Lambda均阴性。电镜:系膜扩张,系膜区可见较多电子致密物沉积,无明确亚结构,内皮下、上皮下及基膜内未见电子致密物,足细胞节段性足突融合;基膜质地正常、平均厚度为(263±48) nm。肾小管上皮病变不明显。例2:肾脏穿刺组织21个肾小球,其中1个球性硬化,2个节段性硬化(图1、2),未见新月体或坏死,5个节段性轻度系膜增生(系膜基质增多)(图3、4),其余肾小球未见明显异常。肾小管萎缩及间质纤维化不明显,散在少量单个核细胞浸润。肾小球小动脉及小叶间动脉未见明显异常。免疫荧光:IgA(2+)、C3(+~2+),位于系膜区及节段性副系膜区,其余均阴性(图5、6)。电镜:节段性系膜增生,系膜区见电子致密物沉积,内皮下、上皮下及基膜内未见电子致密物,足细胞节段性足突融合;基膜平均厚度为(236±2) nm,偶见节段性分层(图7~9)。肾小管病变不明显。2例肾脏穿刺组织免疫组化标记FN于肾间质中等强度阳性,肾小球及肾小管上皮细胞阴性(图10)。

图1 PAS染色示肾小球局灶节段性硬化,肾小管未见萎缩及肾间质纤维化,间质散在少量单个核细胞浸润 图2 PASM染色示肾小球局灶节段性硬化,基膜淡染 图3 PAS染色示肾小球毛细血管襻结构完整,系膜轻度节段性增生 图4 PASM染色示肾小球毛细血管襻结构完整,系膜轻度节段性增生,基膜淡染 图5 肾小球免疫荧光IgA系膜区及节段性旁系膜区阳性,团块及颗粒状 图6 肾小球免疫荧光C3系膜区阳性,弱于IgA阳性强度 图7 电镜低倍镜下肾小球节段性轻度系膜增生,足突节段性融合,基膜薄 图8 系膜区可见电子致密物,未见亚结构 图9 肾小球基膜均质变薄 图10 免疫组化染色FN蛋白在肾小球及肾小管内阴性,鲍曼囊及肾间质中等阳性,EnVision法
2.3 基因变异特点6例样本中2例存在FN1(NM212482.1):c.3289G>A(p.Glu1097Lys)、FN1(NM212482.1):c.4616C>G(p.Ser1539Cys)变异,其余样本未检测出FN1基因变异。FN1 21号外显子错义突变致编码蛋白1 097位谷氨酸替代为赖氨酸,29号外显子错义突变致编码蛋白1 539位丝氨酸替代为半胱氨酸。依据数据库频率信息及各分析平台预测结果,且依据ACMG指南均判断为可能有害变异。
例1还存在NUP160(NM015231.1):c.160G>A(p.Glu54Lys)变异,1号外显子错义突变导致编码蛋白54位谷氨酸替代为赖氨酸,依据数据库频率信息及各分析平台预测结果,ClinVar未查到该变异的致病信息,且依据ACMG指南判断为意义未明变异。例2还存在ITGA3(NM002204.2):c.3046-4G>A变异,依据数据库频率信息,功能软件DANN评分0.918,dbscSNV评分0.032,且依据ACMG指南判断为意义未明变异(表1,图11)。

表1 FN1、NUP160、ITGA3基因变异
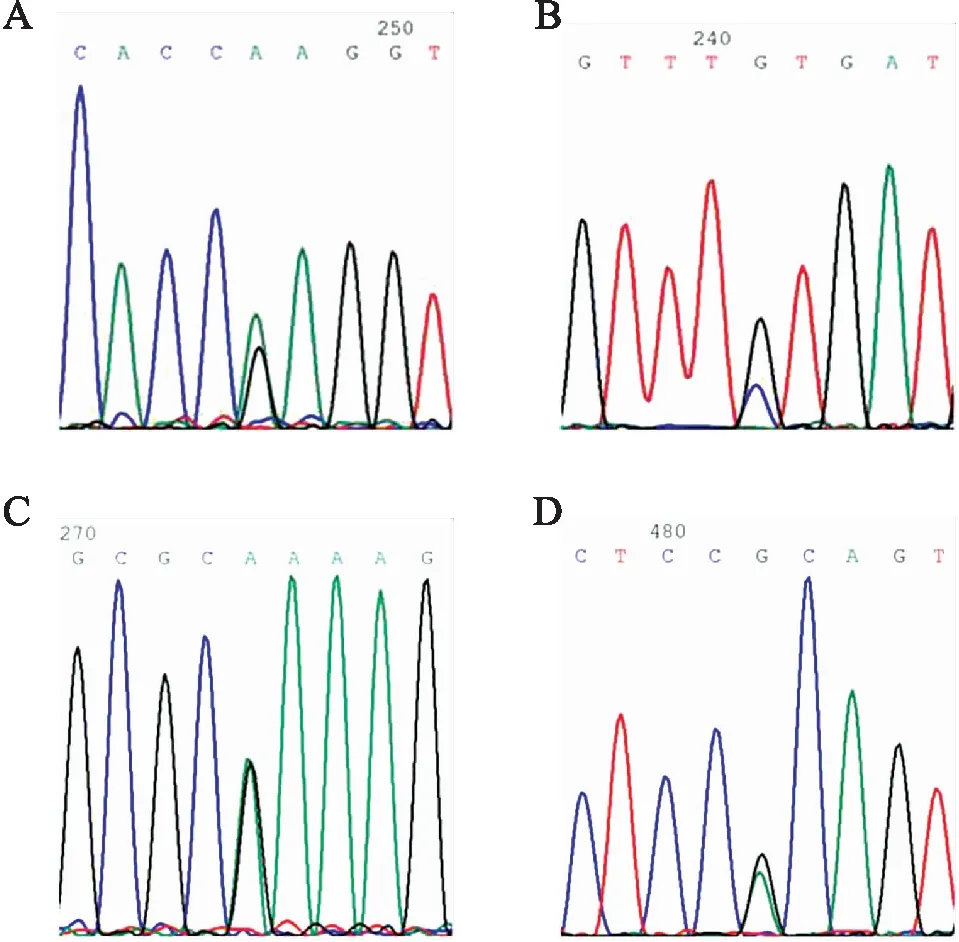
图11 Sanger测序峰图:A.FN1 c.3289G>A;B.FN1c.4616C>G;C.NUP160c.160G>A;D.ITGA3c.3046-4G>A
2.4 氨基酸链空间结构及蛋白结构域、功能预测FN1:c.3289G>A(p.Glu1097Lys),变异位点对应FN1蛋白第6位的FNⅢ型结构域,临近肝素结合结构域位点;FN1:c.4616C>G(p.Ser1539Cys)变异位点对应FN1蛋白第10位的FNⅢ型结构域,位于整合素结合结构域位点,参与细胞间的黏附功能(图12A)。从氨基酸空间结构来看,与既往已报道的明确致病、可能致病、良性非致病的空间位置明显不同(图12B、C)。从蛋白互作网络分析,FN1与ITGA3蛋白存在紧密相关的作用,而NUP160相对独立,推测伴薄基膜改变的IgA肾病受参与基膜结构FN1、ITGA3和足细胞NUP160两条不同的通路影响(图12D)。

图12 A.采用IBS.Biocuckoo软件作图构建FN1蛋白结构域分布图;B、C.FN1变异位点在氨基酸链空间结构:SwissModel数据库显示FN1两个变异位点对应的编码氨基酸位点(粉色)Glu1097及Ser1539在氨基酸链空间结构分布不同于数据库中既往报道过的致病性、可能致病性(红色)及良性非致病性(绿色)位点;D.String数据库显示FN1、ITGA3、NUP160蛋白相互作用关系:FN1与ITGA3存在密切相互作用,NUP160与这两个蛋白不存在相互作用
3 讨论
FN1基因位于染色体2q35,含46个外显子,编码纤连蛋白2 477个氨基酸,是多功能糖蛋白,分血浆型和细胞型,可连接细胞外基质与细胞内肌动蛋白细胞骨架并与调节特异性信号转导级联的细胞表面异二聚体整合素家族中的多个成员作用,也可结合其它细胞外基质糖蛋白、胶原、硫酸乙酰肝素蛋白多糖。功能性纤连蛋白由羧基末端二硫键相连的两个220~250 kDa亚基构成二聚体,每个单体由三种不同的同源重复结构域组成,称为Ⅰ、Ⅱ和Ⅲ型。细胞型纤连蛋白以多聚体形式装配成丝状纤维而存在于细胞外基质的疏松结缔组织、基膜和细胞表面,其多种功能的实现依赖于前体mRNA复杂的时序和细胞种类特异性选择性剪切,尤其是在Ⅲ型结构域。
既往对肾小球基膜中是否存在FN1蛋白意见不一,近年蛋白组学及生物信息学技术鉴定了更多参与细胞外基质及基膜的成分[10-11],FN1也被包括在内,这在鼠及人类中均得以证明。肾小球基膜发育成熟存在层级装配程序,胶原Ⅳα345构成基膜的主要支架,laminin 521和足细胞及内皮细胞表面受体Integrinα3β1和蛋白聚糖α-dystroglycan结合,联系细胞间相互作用。肾小球基膜的发育成熟需要足细胞和内皮细胞共同作用,而FN1对内皮细胞结构的定位及功能起重要作用[12-13]。
迄今为止,FN1在肾小球基膜中的作用机制尚未见详细报道,已知的FN1基因变异导致纤连蛋白肾小球病[14],本组病例的临床表现和病理学形态不同于纤连蛋白肾小球病,而且均具有皮疹,这是在FN1变异疾病的临床表现方面首次被提及,提示FN1:c.3289G>A及c.4616C>G变异在伴薄基膜改变的IgA肾病的肾外表现方面要关注皮肤病变。有研究提示纤连蛋白肾小球病中主要为血浆型pFN,所报道的基因变异位点多数位于肝素结合结构域,少数位于整合素结合结构域。本组病例为伴薄基膜改变的IgA肾病,推测可能和变异所影响的氨基酸链空间结构不同有关,此变异位点影响更多的可能是细胞型cFN。此外,Kliewe等[15]发现FN1参与足细胞对机械应力的感受及调节,涉及ITGA3、ITGB1等整合素及黏着斑蛋白。既往也有一篇关于FN1基因变异致薄基膜病的家系验证性报道[16],也有学者利用生物信息学分析技术筛选到IgA肾病中包括FN1在内的关键基因[17]。因此,FN1在纤连蛋白肾小球病、足细胞病、糖尿病等代谢性肾病、伴薄基膜改变的IgA肾病中存在不同的作用机制。
有报道称ITGA3变异引起基膜及足细胞改变而致蛋白尿[18]。本组例2的ITGA3:c.3046-4G>A变异虽然在剪切功能预测软件中注释为可能并不被影响,但鉴于ITGA3对肾小球基膜的形成及足细胞的黏附作用与FN1密切相关,如有可能仍需进一步行功能学实验加以验证。多种核孔复合体基因变异与激素抵抗型蛋白尿的发生有关。NUP160参与组成核孔复合体Y型结构,起支撑作用,此基因变异会影响环内部其它构成蛋白间结合稳定性从而改变核孔复合体结构和细胞核形态,而NUPs致蛋白尿发生的机制还在探索中,有报道显示NUP160也可影响足细胞中细胞骨架调节成分CdC42的活性[19]。Wang等[20]报道敲低NUP160促进足细胞凋亡、自噬,Nephrin、Podocin和CD2AP表达降低,推测敲低NUP160可促进足细胞迁移,影响细胞骨架及与基膜的黏附。本组例1早期即表现为轻中度蛋白尿,推测除FN1基因外,NUP160基因变异可能也具有协同作用。
本实验首次发现伴薄基膜改变的IgA肾病中存在FN1基因的可能致病性变异,同时推测NUP160、ITGA3基因变异具有引起此类疾病蛋白尿发生的协同作用,为临床FN1基因变异所致疾病表型谱增加了伴皮肤病变及薄基膜改变的IgA肾病这一新的类型,也提示伴薄基膜改变的IgA肾病中存在非胶原Ⅳ基因变异,为此类患者的临床管理及基因变异检测提供依据。
猜你喜欢
湖北农业科学(2022年11期)2022-07-18
天津医科大学学报(2021年4期)2021-08-21
实用肿瘤学杂志(2020年4期)2020-12-08
支部建设(2020年15期)2020-07-08
百科知识(2015年18期)2015-09-10
医学研究杂志(2015年9期)2015-07-01
小星星·阅读100分(高年级)(2015年4期)2015-05-26
医学综述(2011年12期)2011-12-09
亚热带农业研究(2011年3期)2011-09-29
老友(2010年9期)2010-09-16
