
环十二酮肟的结构表征
2022-12-27 02:53许醉笑
分析仪器 2022年6期
许醉笑
(江苏赛孚士生物技术有限公司,泰州,225300)
环十二酮肟是尼龙12生产过程中重要的中间体,酮肟于393±5K下在98%硫酸中能发生分子重排反应生成十二内酰胺(尼龙12单体)[1]。肟类衍生物是新农药研究的一个热点,在新农药的研究中曾发现,环十二酮肟及其衍生物是一类具有农药活性的化合物,例如α-苯磺酰基环十二酮肟酯[2]和α-甲硫基环十二酮肟酯[3]具有良好的除草活性,α-氧代环十二酮肟醚[4],α-甲硫基环十二酮肟醚[5]具有良好的杀菌活性。可见环十二酮肟是重要的化工中间体,有着广泛的应用前景。目前关于环十二酮肟及其衍生物的研究大部分集中在合成与应用方面,而在结构表征、含量测定方面却鲜有报道。因此在本研究中,对以环十二酮为原料制得的环十二酮肟通过红外光谱、质谱、1H-NMR、13C-NMR等手段进行结构表征,确定其为环十二酮肟,并首次采用1H-NMR测定了环十二酮肟含量,建立了环十二酮肟的表征方法。为今后环十二酮肟及其衍生物的结构研究、定量分析提供参考。
1 试验部分
1.1 仪器与试剂
iS50 FTIR红外光谱仪,美国Thermo公司;AVANCE III HD-500MHz核磁共振仪,德国Bruker公司;GCMS-QP2010气质联用仪,日本岛津公司。
氘代氯仿;环十二酮,分析纯;环十二酮肟,自制。
1.2 试验方法
1.2.1 红外
将待测样品的白色细粉,采用ATR方式,分辨率4cm-1,扫描 32次,扫描范围 400~4000 cm-1,进行红外扫描测定。
1.2.2 核磁
称取约20mg待测样品于核磁管内,加入0.5mL氘代氯仿溶解后测定。氢谱采集参数如下:共振频率500.386MHz,测定温度295k,脉冲序列zg30;弛豫时间(D1)1.0s;脉冲宽度(P1)14.8µs; 扫描次数(NS)16次;空扫数(DS)两次;中心频率(O1P)为5.0ppm;谱宽(SW)为10.0ppm。碳谱采集参数如下:共振频率125.834MHz,测定温度295k,脉冲序列zgpg30;弛豫时间(D1)2.0s;脉冲宽度(P1)9.56µs; 扫描次数(NS)1024次;空扫次数(DS)4次;中心频率(O1P)为100ppm;谱宽(SW)为220ppm。
1.2.3 质谱
称取1mg待测样品粉末于专用样品管中,将样品管小心放入固体进样杆中,采用直接进样方式,离子源:EI,电离电压:70eV,离子源温度:250℃。质量范围为m/z33~260。
2 结果讨论
2.1 质谱分析
图1为待测样品的质谱图,m/z197为分子离子峰,这与环十二酮肟的分子量一致,由于环较大,分子离子峰不强。m/z180为酮肟脱羟基形成,m/z165为酮肟氢转移脱NHOH形成。m/z154为环开裂后,脱去C3H7游离基形成;m/z140为环开裂后,脱去C4H9游离基形成;m/z126为环开裂后,脱去C5H11游离基形成;m/z98为环开裂后,脱去C7H15游离基形成。m/z113为环开裂后,脱去C6H12游离基形成;m/z99为环开裂后,脱去C7H14游离基形成。基峰(m/z73)是经α裂解羟基转移后失去C7H14CN形成的C4H8OH+,m/z86为失去C6H13CN形成的C5H9OH+。脱去C7H14游离基形成。m/z41、55、95、109分别为C3H5+、C4H7
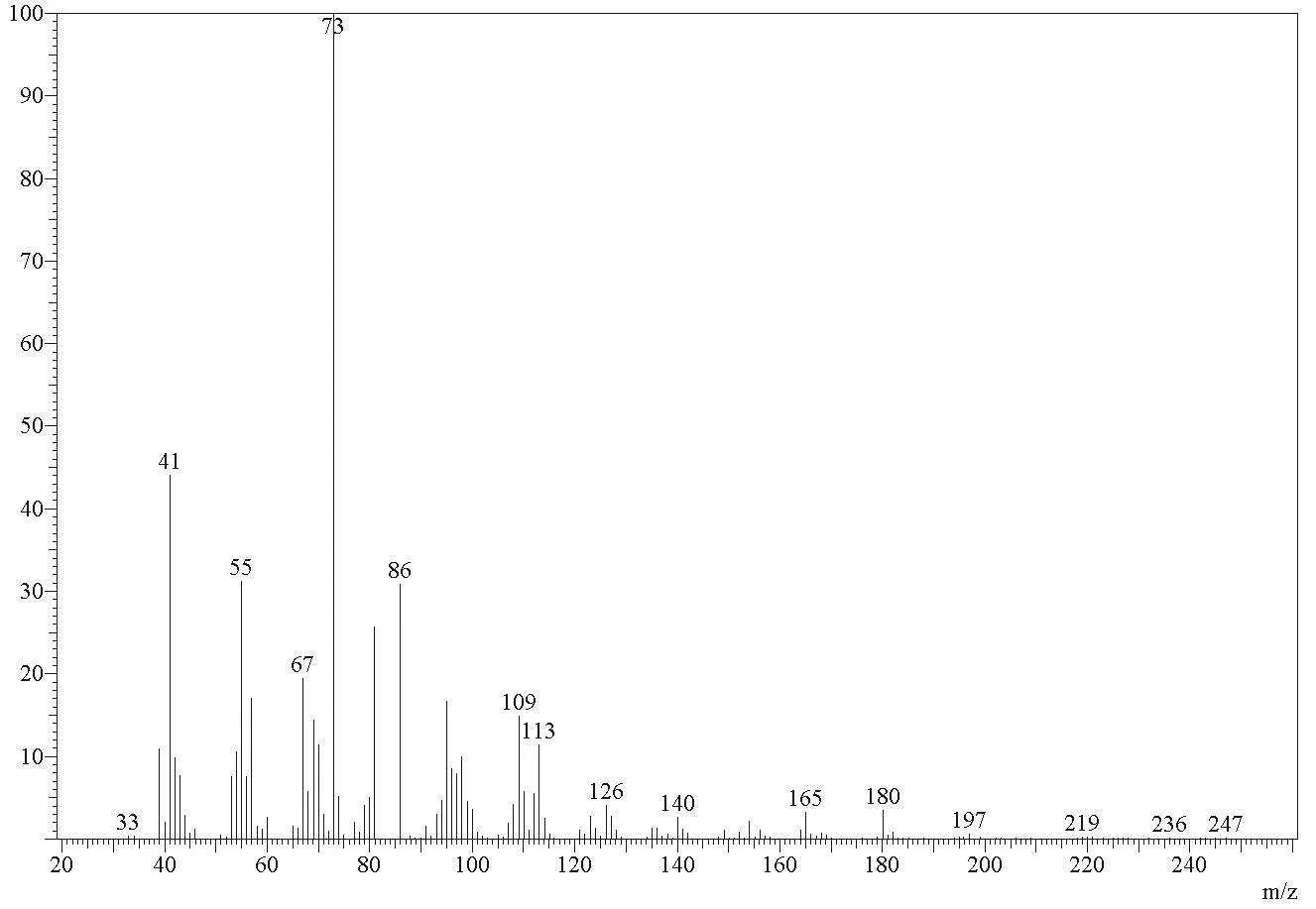
图1 待测样品质谱图
+、C7H11+、C8H13
+,是环开裂形成的不饱和烃。m/z67为C3H5C≡N+,是由m/z180的离子破裂而成。
据报道[6]环十二酮肟的质谱裂解反应主要为酮肟的裂解及环的裂解两种方式。环的裂解形成两个系列的离子,其一为酮肟的氮原子引发α位开裂,邻近位置上的氢转移,脱去CnH2n+1游离基,形成CnH2n-1CNOH+离子;另一系列为CnH2n-CNOH+,是由环开裂后,不经氢转移直接脱去CnH2n形成的。该样品的质谱裂解与文献报道的一致,其中m/z180和m/z165为酮肟裂解,m/z154、m/z140、m/z126、m/z113等为环的裂解,符合环十二酮肟的裂解机理,可以初步推测该样品为环十二酮肟。
2.2 红外分析
图2为待测样品红外光谱图,其中3235.85cm-1吸收峰为肟类C=N-OH的-OH伸缩振动,1671cm-1吸收峰为肟类C=N-OH的-C=N伸缩振动,2993.51 cm-1、2845.88 cm-1为亚甲基的C-H伸缩振动,1467.88 cm-1为亚甲基的C-H变形振动。上述红外基团特征吸收峰与环十二酮肟的结构吻合。
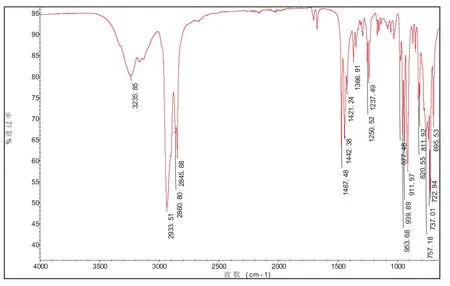
图2 待测样品红外光谱图
2.3 核磁分析
在核磁共振碳谱中,化学位移161.26ppm为与肟羟基相连的碳,化学位移30.37ppm 为C2位上的-CH2,其他(C3~C11)位上的-CH2化学位移在30以下。
图3为待测样品核磁共振氢谱,从氢谱中可以看出,该样品含有5类不同质子氢,其中大部分在高场为环十二烷基氢。化学位移1.329~1.387ppm处的多重峰为C7~C10位上的7个-CH2;化学位移1.603~1.667ppm处的多重峰为C3和C11位上的2个-CH2;受肟基的影响C2和C12位上的两个-CH2向低场移动,化学位移在2.281~2.453ppm之间。由于环十二酮肟的空间构型不对称,C2位上的-CH2受同侧肟羟基氧原子的去屏蔽作用,与C12位上的-CH2相比,化学位移移向低场,即化学位移2.4ppm处的三重峰为C2位上的-CH2,化学位移2.3ppm处的三重峰为C12位上的-CH2。低场化学位移8.3ppm处的宽峰为肟基上的羟基氢。5类质子氢的积分强度比为0.98:2.12:2.00:4.05:14.26,与环十二酮肟1:2:2:4:14相符,可以推测该样品为环十二酮肟。
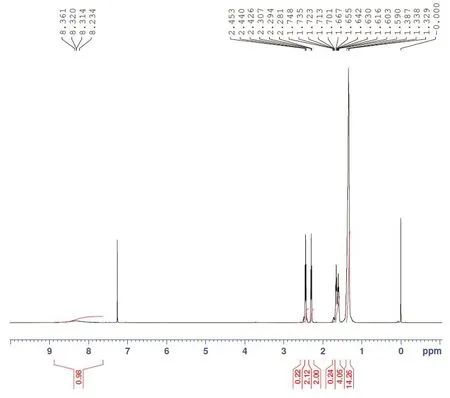
图3 待测样品核磁共振氢谱
图4为待测样品核磁共振碳谱,化学位移161.26ppm为与肟羟基相连的碳,化学位移30.37ppm为C2位上的-CH2,其他(C3~C11)位上的-CH2化学位移在30以下。
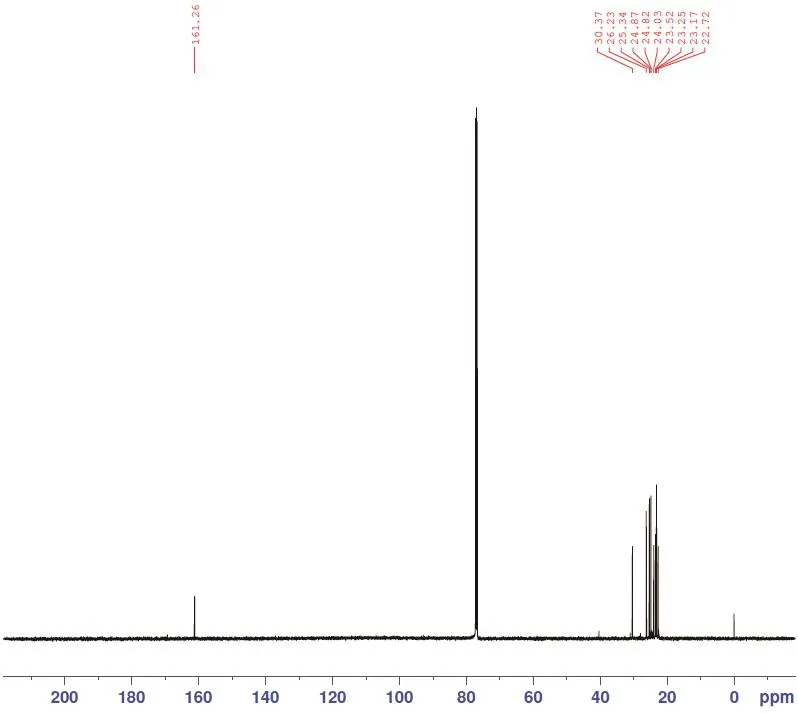
图4 待测样品核磁共振碳谱
结合上述质谱、红外、核磁氢谱及碳谱结果分析,可以推断该样品为环十二酮肟,表明以环十二烷为原料所制得的产物为目标产物。
由图3环十二酮肟氢谱可以看出,该样品中含有少量杂质,推测为少量的原料残留(即环十二酮)。图5为环十二酮的氢谱图,化学位移2.453~2.477ppm处的多重峰为C2和C12位上的两个-CH2,化学位移1.701~1.725ppm处为C3和C11位上的两个-CH2,化学位移1.267~1.306ppm处的多重峰为C4~C10位上的7个-CH2。对比发现,环十二酮肟氢谱中化学位移1.701~1.725ppm处为环十二酮C3和C11位上的两个-CH2,可用该基团测定两类物质的相对含量。计算公式如下,该样品中环十二酮肟含量为94.41%。
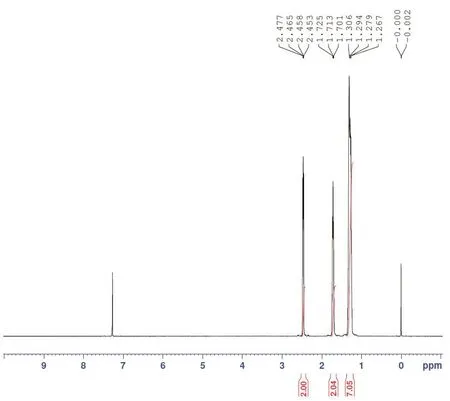
图5 环十二酮核磁共振氢谱
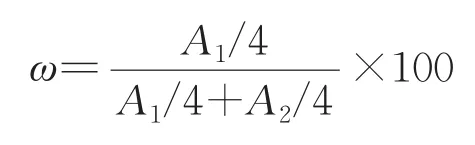
其中ω为环十二酮肟相对含量,A1为环十二酮肟C3和C11位上的两个-CH2的积分强度,A2为环十二酮C3和C11位上的两个-CH2的积分强度。
2.4 含量计算
目前,关于环十二酮肟含量测定的研究未见报道。而与之结构相似的环己酮肟多采用间接测定法,如在酸性介质中,将环己酮肟水解成羟胺,利用羟胺的显色反应,通过分光光度法对其进行测[7];也有文献采用高效液相色谱法[8]、气相色谱法[9]测定环己酮肟的含量。以上方法成熟,但分光光度法需要繁琐的前处理,色谱法需要标准品制定标准曲线,检测时间长。核磁共振氢谱因其共振峰的积分值与被测组分的含量成正比而可用于定量分析[10-12],该定量法具有不破坏样品、所需样品量小、一般不需前处理、无需对照品、专一性强、检测速度快等优点。核磁共振氢谱定量主要有3种方法:内标绝对测定法、外标绝对测定法、相对含量测定法[13]。在本研究中采用相对含量测定法。
3 结论
在本研究中,通过红外光谱、质谱、1H-NMR、13C⁃NMR等手段对以环十二烷为原料所制得的产物进行结构表征,确定其为环十二酮肟,并通过1H‒NMR测定了环十二酮肟相对含量,建立了环十二酮肟的定性定量方法。为今后环十二酮肟及其衍生物生产制备提供参考。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中国抗生素杂志(2022年7期)2022-08-18
科学技术创新(2022年36期)2022-02-01
中国油脂(2019年1期)2019-01-23
中成药(2018年2期)2018-05-09
新乡学院学报(2016年6期)2016-12-01
中国塑料(2016年12期)2016-06-15
云南地质(2015年3期)2015-12-08
中国当代医药(2015年8期)2015-03-01
应用化工(2014年8期)2014-08-08
