
细胞培育肉种子细胞永生化诱导研究
2023-01-12 05:54杨峰李莹莹王守伟刘文婷李石磊
中国食品学报 2022年12期
杨峰,李莹莹,王守伟,刘文婷,李石磊
(中国肉类食品综合研究中心 北京食品科学研究院 北京 100068)
细胞培育肉也被称作培养肉、人造肉、清洁肉等[1-2],是利用动物细胞体外培养的方式控制其快速增殖、定向分化,收集加工而成的一种新型肉类食品[3-4],是基于生物工程技术生产的肉类产品。该类肉品因可以绕开动物饲喂而可持续地为人类供应真实动物蛋白,被认为是最有可能解决未来人类肉品生产和消费困境的方案[5]。相对于植物蛋白素肉,培育肉以其可为人类提供真实动物蛋白而被广泛看作是最具商业价值潜力的肉类替代物[6],在生产技术、发展理念和存在意义等方面具有较强的先进性。与传统肉类生产方式相比,细胞培育肉生产占用的土地面积将减少95%,水消耗量将减少75%,温室气体排放量将减少87%,能源消耗将减少45%,这将从技术层面助力肉类产业实现“双碳目标”[7-8],推动肉类食品产业绿色可持续发展。
目前细胞培育肉的生产主要包括细胞选择和细胞库建立、规模化增殖、细胞分化、细胞收获、终产品加工[9]。种类丰富、高活力的种子细胞是生产细胞培育肉的源头,种子细胞的获取也是生产细胞培育肉的关键环节[10]。目前开展的细胞培育肉研究,种子细胞主要通过从牛[11-13]等动物中提取、分离获得高纯度的肌肉干细胞,然而,这些细胞活化后分化为成肌细胞即失去干性,无法永久性持续增长,细胞传代能力仍然面临着诸多挑战[14-15]。另外,持续从动物机体获取细胞也违背了动物福利[16]。细胞培育肉技术的快速发展推动了细胞培育肉的工业化进程,也面临着大量种子细胞制备的需求,因此亟待构建高效、可持续的种子细胞的制备方法。
细胞永生化技术[17]可以使得细胞获得持续生长的增殖能力,长期传代,并具有无限增殖的生长特性,克服一般原代细胞有限次传代后即发生衰老和死亡的情况。成体细胞的永生化,包括端粒酶逆转录酶转染[18-19]、基因敲除类恢复性诱导[20]、四小吉奎琳氧化法以及黄曲霉素刺激法[21-22],然而,目前仅限于基础研究,现存的永生化细胞系多为小鼠或人源细胞,无法直接应用于细胞培育肉体系,更不能用于食品科学的研究。本研究利用猪成肌细胞,通过端粒酶逆转录酶进行诱导载体的构建,核转染到细胞后分别通过免疫荧光标记、定量PCR、染色体核型分析等方法进行细胞诱导验证,以期实现猪源永生化成肌细胞的诱导,构建稳定性强的猪成肌细胞永生化诱导载体,为细胞培育肉种子细胞制备提供更高效的技术手段,为细胞培育肉工业化生产种子细胞获取打下基础。
1 材料与方法
1.1 材料与试剂
猪肌卫星细胞取自新生猪胎儿大腿肌肉,通过组织贴壁法进行培养传代;GatewayBP Clonase II Enzyme Mix,GatewayLR ClonaseTMII Plus Enzyme Mix,Lipofectamine 2000 Reagent,Taq DNA Polymerase,100 bp DNA Ladder,dNTP Mix,GeneRuler,GeneRuler TM 1 kb DNA Ladder,美国赛默飞世尔科技有限公司;PrimeSTAR HS DNA Polymerase,日本Takara 公司;Sanprep 柱式胶回收试剂盒,生工生物工程(上海)股份有限公司;RNA 提取试剂盒,质粒小提试剂盒,DH5α 感受态细胞、无内毒素质粒大提试剂盒,天根生化科技(北京)有限公司;DMEM 培养基,Ampicillin LB琼脂平板,云舟生物科技(广州)股份有限公司;限制性内切酶,胰酶,美国NEB 公司;胎牛血清,细胞培养皿,美国康宁公司;Lenti-X p24 Rapid Titer Kit,美国Clontech 公司;EP 管,美国Axygen公司;Triton X-100,牛血清白蛋白,山羊血清,嘌呤霉素,北京索莱宝科技有限公司;其它试剂为国产分析纯级。
1.2 仪器与设备
PCR 仪、电泳仪、凝胶成像系统,美国Bio-Rad 公司;水平电泳槽,北京六一生物科技有限公司;倒置荧光显微镜,日本奥林巴斯公司;流式细胞仪,美国贝克曼公司;NanoDrop8000,美国赛默飞世尔科技有限公司;台式高速冷冻离心机、冰箱(-80 ℃)、CO2培养箱、恒温摇床、生化培养箱,上海一恒科学仪器有限公司;台式冷冻离心机,美国艾本德有限公司;涡旋混合器,德国IKA 公司;恒温水浴锅,上海比朗仪器;震荡培养箱,上海旻泉仪器有限公司;冰箱(-20 ℃),中科美菱低温科技股份有限公司;超纯水仪,美国Millipore 公司。
1.3 试验方法
1.3.1 永生化诱导载体的构建 猪成肌细胞永生化诱导载体构建流程如下:
引物设计,PCR 克隆目的片段→目的片段切胶回收→BP 反应构建中间载体→中间载体测序鉴定→LR 反应,转化及终载体阳性克隆鉴定。
1.3.1.1 BP 反应构建pDown-TERT[NM_198253.3]中间载体 根据BP 反应原理,设计引物序列:attB site+特异性引物序 列:TERT BP-F:5'-GGGGACAAGTTTGTACAAAAAAGCAGGCTATGC CGCGCGCTCCCCTERT;BP-R:5 -GGGGACCAC TTTGTACAAGAAAGCTGGGTTCAGTCCAGGATG GTCTTGAAGTCTG。PCR 反应体系:5×Primer STARTM Buffer,10 μL;dNTP Mixture(10 μmol/L),4 μL;引物-F(10 μmol/L )、引物-R(10 μmol/L)、模板DNA,各1 μL;Primer STARTMHS DNA Polymerase,0.5 μL;总体积50 μL。PCR 反应程序:98 ℃,3 min;98 ℃10 s,60 ℃10 s,72 ℃60 s,循环次数均为30 次;72 ℃5 min。PCR 反应结束后,进行琼脂糖凝胶电泳,从琼脂糖凝胶切下含目标片段的胶块,通过Sanprep 柱式胶回收试剂盒进行DNA 回收,将DNA 置于-20 ℃保存备用。
参考质粒小提试剂盒说明书,制备该载体构建过程中使用到的各种骨架载体DNA。BP 反应体系:attB-PCR 产物,75 ng;pDONR 221 骨架载体,75 ng;BP ClonaseTMII Enzyme Mix,1 μL;TE Buffer,pH 8.0,加至4 μL。BP 反应条件:25 ℃孵育1 h;反应后,加1 μL 蛋白酶K,37 ℃孵育15 min 终止BP 反应。
从-80 ℃中取出感受态细胞,冰上解冻;在超净工作台中,取5 μL BP 反应产物加入100 μL感受态细胞中,轻弹混匀;冰浴30 min;42 ℃热激90 s;冰浴2~3 min;加入250 μL LB 培养基;250 r/min,37 ℃,摇菌培养(复苏)1 h 左右;将复苏的菌液涂布到含有Kana 抗生素的平板上,37 ℃倒置培养过夜。随机挑取3~6 个单菌落,将菌体涮洗在无菌0.2 mL 的无菌EP 管中,取1 μL 做菌落PCR的模板,剩余作为接种培养细菌抽提质粒DNA 的菌种。
菌落PCR 反应体系:模板1 μL,dNTP Mixture(2.1 mmol/L)0.8 μL,测序引物-F1(10 μmol/L)0.5 μL,测序引物-R(10 μmol/L)0.5 μL,10 ×Buffer 1 μL,Taq DNA Polymerase 0.5 μL。菌落PCR 反应程序:94 ℃3 min;94 ℃30 s,60 ℃30 s,72 ℃1 min/1~2 kb,循环次数均为25 次;72 ℃5~10 min。菌落PCR 结束之后,加入6×loading buffer 后电泳,参照DNA Ladder 挑选出能扩增出目标长度的克隆,接种到LB 培养基中,37 ℃250 r/min 培养过夜,随后抽提小提质粒DNA 送测序,采用Sequencher 软件比对返还的测序结果和标准序列,测序正确的入门载体可以进入下一步LR反应构建最终载体。
1.3.1.2 LR 反应构建pLV [Exp]-EGFP:T2A:Puro-CMV>TERT[NM_198253.3]最终载体 通过LR 反应将目的序列重组到最终骨架载体,获得含有目的序列的重组表达载体。首先进行LR 反应。LR 反应体系:pUP-CMV 10 ng,pDown-TERT[NM_198253.3] 10 ng,pLV[Exp]-EGFP:T2A:Puro 50 ng,LR clonase 1 μL,TE buffer 加至5 μL 终体系。LR 反应条件:25 ℃孵育3 h。反应结束后加入1 μL 蛋白酶K 37 ℃孵育15 min 终止反应。之后对反应产物转化感受态细胞,菌落PCR,琼脂糖凝胶电泳,接种培养细菌并抽提质粒DNA(方法同1.3.1.1 节)。采用NEB 的限制性内切酶对最终载体的质粒DNA 进行酶切,酶切反应结束后,琼脂糖凝胶电泳并参照DNA Ladder 进行分析,能且仅能切出特定长度的DNA 条带的载体ApaLI+EcoRV:936,660,3 985,1 246,5 356,即为正确的最终载体。
1.3.1.3 慢病毒包装 对终载体进行质粒DNA大提(方法参照质粒提取试剂盒说明书),通过脂质体法转染HEK293T 细胞及收毒,即:将HEK293T 细胞培养至融合度为80%~90%;更换新培养基1 h 后加入Opti-MEMI 培养液继续培养;分别混合A、B 液,其中A 液为1.5 mL Opti-MEM 与4 μg DNA(分别为包装质粒SL3、SL4、SL5 以及目的基因质粒),将质粒加入Opti-MEM中,然后用枪头吹打混匀;B 液为1.5 mL Opti-MEM 与40 μL Lipofectamine 2000,用枪头吹打混匀,室温静置孵育5 min;将A 液加入B 液中,用枪头吹打混匀,室温静置孵育20 min;将孵育后的转染复合物缓慢逐滴加入培养细胞的100 mm培养皿中,水平轻微晃动使其分布均匀,37 ℃,5%CO2培养,转染6 h 后更换培养基为含10% FBS的DMEM 培养基,继续37 ℃,5%CO2培养;转染48 h 后收集培养基上清存放于4 ℃,添加同体积新鲜的含10% FBS 的DMEM 培养基继续培养,过24 h 再收集1 次培养基上清;将两次收集的培养基上清一起进行浓缩,4 ℃,1 000×g 离心30 min,离心所得上清用0.45 μm 滤器过滤去除细胞碎片;将滤液加入离心管中,4 ℃50 000×g 离心2 h,弃上清,每管沉淀用200 μL HBSS 缓冲液重悬慢病毒颗粒,分装后置于-80 ℃保存,得到的病毒可用于转导细胞。
慢病毒纯化:将200 μL 病毒浓缩液加入离心管中,上层加入1.5 mL 20%蔗糖溶液,平衡后4℃50 000×g 离心2 h;离心去上清,收集沉淀病毒颗粒,每管沉淀用200 μL HBSS 缓冲液重悬,分装后冻存于-80 ℃,然后,采用ELISA 法测定慢病毒滴度。
1.3.1.4 细胞转导 转导前1 d,将一定数量的靶细胞接种至新的培养皿中(转导时靶细胞的融合度在30%~50%),置于37 ℃、5% CO2的培养箱中培养18~20 h;转导当天,在冰上溶解冻结的病毒液,溶解后轻柔吹打混合病毒颗粒,去适量病毒至培养基中,轻柔混匀,尽可能地减少培养基使用量(100 μL/cm2),将培养板置于37 ℃、5% CO2培养箱中过夜培养;3~4 h 后取出培养基,换液,洗出含有病毒的培养基,加入新鲜的完全培养基,置于37 ℃、5% CO2培养箱中过夜培养;每24 h 换液1次,在荧光显微镜下观察绿色荧光表达情况。
将表达荧光的细胞继续培养,药筛之前进行细胞致死试验。分别在24 孔板中加入终质量浓度为0,1,2 μg/mL 和3 μg/mL 的嘌呤霉素,每组6个平行样本,连续培养3~7 d,测试细胞致死状态,制作致死曲线。选择细胞致死时间4~5 d 的嘌呤霉素浓度,加入表达病毒载体的转导细胞中,连续培养4~5 d,直到培养过程中没有死细胞产生为止,观察载体表达情况。
1.3.1.5 细胞核型分析 在5 mL 培养液中加入50 μL 10 μg/mL 秋水仙素,抑制中期分裂的细胞,在37 ℃、5% CO2继续培养3~4 h,直至细胞核中有圆形发亮的球形为止。将细胞培养液转移到15 mL 离心管中,然后,用PBS(37 ℃温浴)洗涤培养瓶中的贴壁细胞,洗涤2 次。加入0.25%胰酶+0.02%EDTA 消化,3 min 左右,加入细胞培养液终止消化,用弯头吸管轻轻吹打细胞生长面,然后,将细胞悬液转移到上述15 mL 离心管中,1 000 r/min 离心10 min,弃上清液,留约0.5 mL。加入预热(37 ℃)的0.075 mol/L KCl 溶液至5 mL,于37℃水浴低渗处理25 min;低渗结束后加入0.5 mL固定液(V甲醇∶V冰乙酸=3∶1,现用现配),混匀后,迅速离心(1 000 r/min,10 min),弃上清液,留少许液体。加入4 mL 固定液,室温固定20 min,1 000 r/min 离心8 min,弃上清液,留少许液体,重复2次。将离心后的细胞悬浮开,以10~30 cm 高度滴到冰水(0~4 ℃)预冷、倾斜45°的载玻片上,并迅速将滴上的细胞吹开,在酒精灯上轻轻过2~3 遍,细胞面朝上,吸水纸吸去多余液体,室温下晾干,约10~15 min;细胞面朝下,扣在染色玻璃板上,打入PBS(1 ∶10)稀释的吉姆萨染色液,避免打入气泡,染色15 min,蒸馏水冲洗,吸水纸吸去多余液体,10~15 min 后显微镜下观察。
1.3.2 细胞端粒免疫荧光染色 PBST:含0.5%Triton X-100 的PBS 溶液。封闭液:含10%山羊血清的PBST 溶液。二抗稀释液:称取1 g 牛血清白蛋白(BSA),溶于100 mL PBS 中,即1% BSA 的PBS 溶液。取出备用细胞,弃培养基,PBS 洗2 次。加入4%固定液室温固定15min。PBS 洗3 次,每次5 min。用PBST 溶液进行透化,室温10 min。PBS 洗1 次,5 min 加入封闭液,室温封闭30~60 min。弃封闭液,加入一抗(1 ∶200)稀释于封闭液中,37 ℃孵育2 h,1% BSA 的PBS 洗3 次,每次5 min 加入二抗(1∶400)稀释于二抗稀释液中,37 ℃避光放置1 h。PBS 洗3 次,每次5 min。加入终浓度的DAPI(1 ∶10 000)/Hoechst(1 ∶5 000),室温放置5 min。PBS 洗3 次,每次5 min。加入500 μL PBS,在荧光显微镜下拍照或避光保存。
1.3.3 流式细胞术鉴定阳性细胞 将单细胞悬液加入2 mL 圆底离心管中,离心,1 500 r/min,5 min,弃上清液。用1 mL 1×PBS 洗涤1 次,离心。4%PFA 1 mL,4 ℃固定30 min。用1×PBS 1 mL 洗涤1 次,离心。1% Triton-100 1 mL,室温10 min。用1 mL 1×PBS 洗涤1 次,离心。加入用PBA稀释的荧光素标记的抗体200 μL,用移液器轻轻吹打混匀,置冰上孵育30~60 min。离心,弃上清液。加入1 mL 冷PBS,离心洗涤2 次,除去未结合的抗体成分。向细胞中加入500 μL 冷PBS,吹打混匀,置流式管中,4 ℃避光保存,上流式细胞仪。
1.3.4 RNA 提取及荧光定量PCR 用细胞刮刮取细胞,在4 ℃离心机中离心,弃上清,采用RNA提取试剂盒提取RNA,将RNA 洗脱后转入-80 ℃保存,备用。以2 μg 体系逆转录RNA 为单链的cDNA。根据RNA 浓度,构建反应体系,即:RNA,2 μg;oligo DT,1 μL;dNTP,1 μL;总体系15 μL。65 ℃金属浴加热5 min,转至冰上冷却降温。冷却后加入以下反应体系:M-MLV,0.5 μL;RRI,0.5 μL;5×Buffer,4 μL。37 ℃金属浴50 min,70 ℃金属浴10 min。置于-20 ℃保存,备用。通过NCBI Gene bank 查询基因TERT(Gene ID:492280)、Hnrnpc(Gene ID:100626496)、β-actin(Gene ID:414396)序列信息。通过NCBI Primer-BLAST 在线设计引物,所设计引物信息如下:

表1 目的基因引物序列Table 1 Target gene primer sequence
以逆转录后得到的cDNA 为模板,稀释10倍,以20 μL 体系进行RT-PCR 试验。反应体系:2 ×SYBR Green PCR Master Mix,10 μL;PCR Forward Prime,PCR Reverse Prime,各0.6 μL;cDNA,2 μL;DEPC 水,6.8 μL,总体积20 μL。每个试验组设置3 个平行试验。冰上操作,依次加入18 μL MIX 溶液,2 μL cDNA。程序如下:第1 阶段,95 ℃10 min;第2 阶段,95 ℃15 s,60 ℃30 s;第3阶段,95 ℃ 5 min;65 ℃ 5 min;95 ℃ 5 min。
采用2-ΔΔCt法处理、分析试验数据。

式中,R——基因相对表达量;Ct0(GOT)——对照组基因的Ct;Ct0(ref gene)——对照组内参基因的Ct;Ct(GOT)——样品组基因的Ct;Ct0(ref gene)——样品组内参基因的Ct。
2 结果
2.1 端粒酶逆转录酶过表达载体构建结果分析
为了探索细胞培育肉用细胞的永生化诱导,通过脱毒的慢病毒进行过表达载体的构建,将TERT 端粒酶逆转录酶CDs 序列进行PCR 全长克隆。结果显示(图1):TERT 序列CDs 区全长为4 801 bp,载体类型为哺乳动物基因表达载体,启动子为CMV,ORG 开放阅读框为本研究目的基因。同时设置EGFP 荧光表达载体和嘌呤霉素药筛原件,载体抗性为氨苄。载体中设置酶切位点有HpaI、ApaLI、DraIII、ApaLI+XhoI、ApaLI+NdeI、ApaLI+HpaI、ApaLI+DraIII。构建完成的载体通过多引物PCR 验证和测序验证,结果良好,序列无突变发生。

图1 永生化诱导过表达载体图谱及突变检测Fig.1 Immortalization-induced overexpression vector map and mutation detection
2.2 体转导最佳滴度及表达时序验证
为了测定细胞转染最佳转导滴度,首先进行空载体转导试验。分别通过10,20,30 滴度对细胞进行转导,明确20 滴度的转导优势后,通过15,20 和25 3 个滴度进行转导验证,结果表明,转导滴度为25 时转导细胞效率最高,进而将病毒载体通过25 滴度的浓度转导到猪成肌细胞中,在48 h 端粒酶逆转录酶载体开始表达,96 h 后表达量开始升高,转导180 h 后,端粒酶逆转录酶的载体表达达到最高值(图2)。细胞转导效率较高,可进行后续试验。

图2 细胞转导后载体表达时序分析Fig.2 Time series analysis of vector expression after cell transduction
2.3 嘌呤霉素筛选阳性细胞药筛及细胞活力检测
为了去除假阳性细胞,通过嘌呤霉素进行阳性细胞的筛选。首先进行细胞培养,当细胞贴壁24 h 时,以0,1,2 μg/mL 和3 μg/mL 的质量浓度对普通细胞做致死试验,绘制致死曲线。结果表明(图3a):空白对照组中的细胞正常增殖,生长曲线呈现S 型,证实细胞的状态和增殖能力处于正常水平。在添加1 μg/mL 嘌呤霉素的组别中,前3 d细胞致死趋势较为平缓,从第3 天开始细胞致死速度加快,而相对2 μg/mL 和3 μg/mL 质量浓度细胞致死效率仍较低,截止第6 天细胞仍未全部死亡,因此,该质量浓度的药筛试验较为漫长,不适合阳性细胞的筛选。在2 μg/mL 组别中,前5 d的细胞致死率趋于一致,到第6 天细胞完全致死,致死速度较为缓和;同时,在5~6 d 细胞全部死亡,符合本研究中细胞的药筛试验。当添加量为3 μg/mL 时,细胞第3 天就全部死亡,说明药筛浓度过高,不利于阳性细胞的增殖。
阳性细胞筛选完成后进行细胞连续传代验证,结果显示:诱导后的第1 代细胞(图3b)的增殖能力显著低于常规同代次的细胞(细胞为猪细胞传代5 代,将冻存1 年的细胞进行诱导处理),表明此时诱导的细胞尚未从细胞转导过程中恢复过来,细胞增殖能力和细胞活力相对较差。空白组细胞进入指数期时,诱导组的细胞尚处于潜伏期,对比空白组,诱导组的细胞没有明确的潜伏期和指数期,同时指数期极不显著,空白组的细胞在第5 天进入平台期后,诱导组的细胞尚未进入指数期。当传代至20 代时(图3c),诱导的细胞增殖能力及活力已经显著恢复,细胞增殖曲线与空白组细胞没有显著差异。当诱导组细胞传代至180 代时(图3d),细胞活力和增殖能力呈下降趋势,差异不显著,同传代20 代的空白对照组细胞相比(常规细胞无法传代至180 代,不能单一变量对比),细胞的增殖能力虽然比空白组差,但是差异不显著。此时诱导组的细胞已经突破生理极限,突破了“Hay flick”界限(细胞最大分裂次数)。由此证明本研究中的细胞永生化诱导取得初步成功。
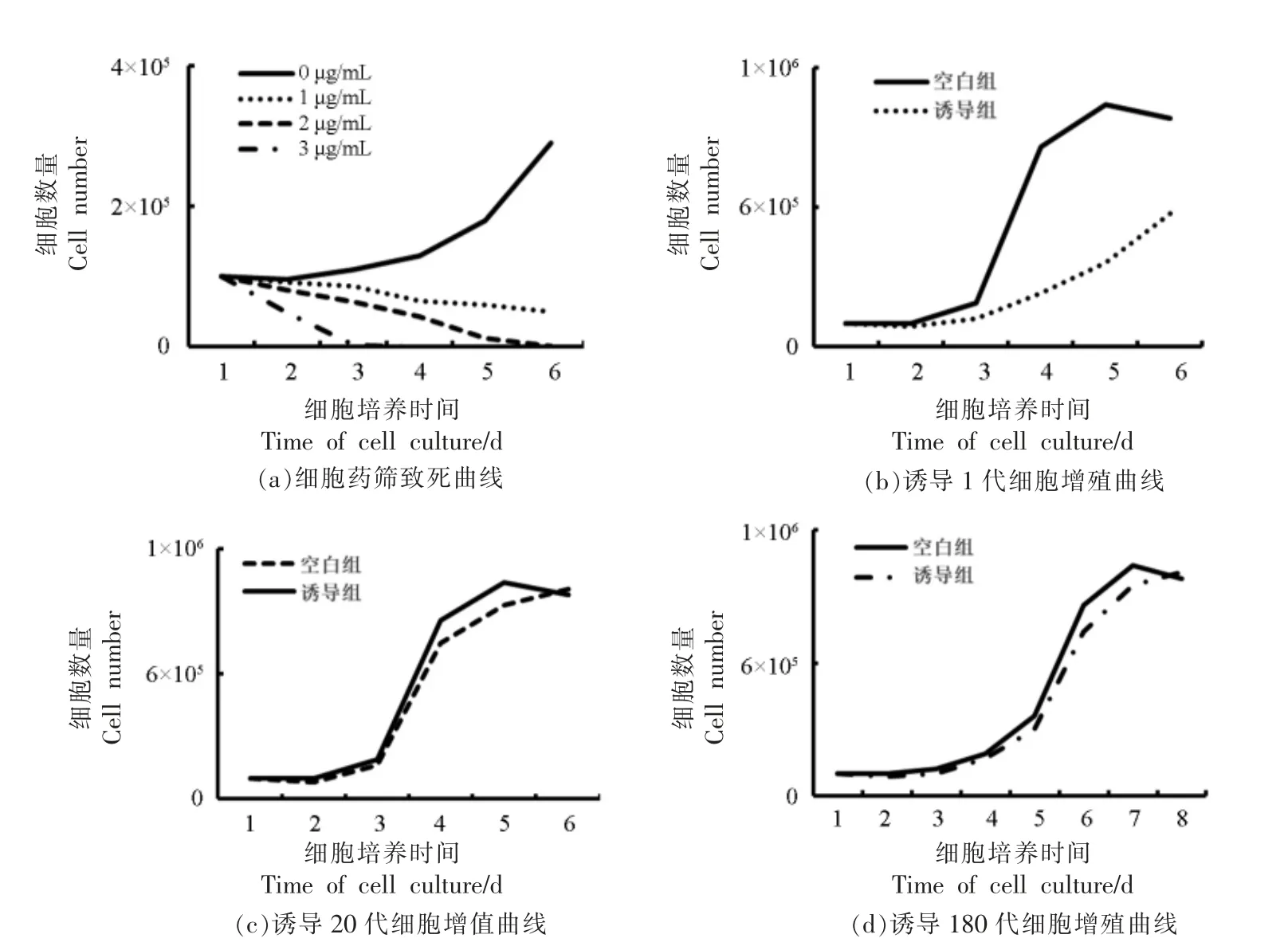
图3 细胞凋亡及增殖曲线Fig.3 Apoptosis and proliferation curves
2.4 流式细胞术纯化阳性细胞及细胞阳性率鉴定
为了检测阳性细胞的药筛效率,分别在诱导细胞加入嘌呤霉素的第2,4,6 天时进行流式细胞术的细胞鉴定。由于有诱导阳性的细胞表达绿色荧光蛋白,因此通过PI 对所有细胞进行染色,通过绿色荧光通道进行细胞分群(图4),结果显示:第2 天时,细胞明显产生分群效应,直方图峰值有两个。同时对细胞进行十字门分群,结果发现,62.11%的细胞为表达绿色荧光蛋白的阳性细胞,36.23%的细胞为未被嘌呤霉素杀死的阴性细胞。当药筛进行到第4 天,直方图结果显示第1 峰值显著降低。对细胞进行圈门,十字门分群后阳性细胞比例为78.92%,显著高于第2 天的62.11%,同时阴性细胞的比例为21.08%,较第2 天显著降低;第6 天时,细胞直方图显示为单峰,阳性细胞比例为100%(存在极少数的阴性细胞在圈门外,比例接近于0),阴性细胞已通过嘌呤霉素全部筛除。通过流式细胞术鉴定细胞药筛效率,一方面可以精准判断阴性细胞的去除效率,减少后期阳性细胞增殖过程中的细胞混合污染问题,另一方面,在筛选过程中当阴性细胞致死后及时更换培养基,避免药筛试验对阳性细胞产生过多的干扰或不确定的影响。

图4 流式细胞术鉴定阳性细胞纯化程度Fig.4 Purification degree of positive cells identified by flow cytometry
2.5 永生化诱导细胞株遗传稳定性鉴定
为了验证成肌细胞永生化诱导后是否产生染色体异变情况,对诱导后传代50 代的细胞进行染色体核型分析(图5)。参照国际家养动物分带核型标准化会议提出的家猪染色体核型分类标准,将猪染色体分为A、B、C、D 组。按照组内染色体相对长度排列,分别标记以1~18 的标号。A 组为第1~5号染色体,亚中着丝粒染色体(sm),其中1 号染色体最长;B 组为6,7 号染色体,亚端着丝粒染色体(st);C 组为第8~12 对染色体,中着丝粒染色体(M),X 染色体为中着丝粒染色体,属于C 组;Y 染色体为所有染色体中最小的,同样是中作私立染色体;D 组为第13~18 号染色体,端着丝粒染色体(T),其中13 号染色体相对长度仅次于1 号染色体。结果显示:1 号染色体中央着丝粒和次缢痕染色深,2 号染色体短臂有4 条深带,长臂有6~7 条深带,分3 个区;3 号染色体着丝粒染色浓,在短臂和长臂的中端各有1 条明显宽阔的浅带;其它染色体从形态、长度和臂比上看均为产生显著的异变,揭示了本研究中永生化诱导未对染色体产生不可预测的异变。
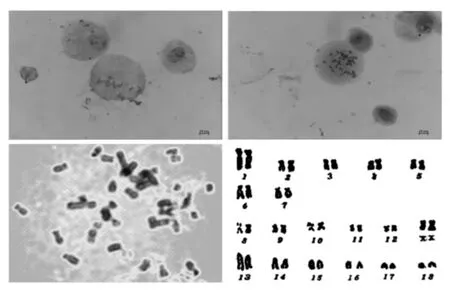
图5 细胞核型分析Fig.5 Karyotype analysis
2.6 端粒酶逆转录酶及端粒四聚体表达鉴定
在染色体复制过程中,细胞每进行一次染色体复制,末端DNA 都会发生丢失。端粒是短的多重复的非转录序列(TTAGGG)及一些结合蛋白组成特殊结构,除了提供非转录DNA 的缓冲物外,它还能保护染色体末端免于融合和退化,在染色体定位、复制、保护和控制细胞生长及寿命方面具有重要作用,并与细胞凋亡、细胞转化和永生化密切相关。当细胞分裂1 次,每条染色体的端粒就会逐次变短一些。构成端粒的一部分基因约50~200个核苷酸,会因多次细胞分裂而不能达到完全复制(丢失),以至细胞终止其功能,不再分裂。严重缩短的端粒是细胞老化的信号。在某些需要无限复制循环的细胞中,端粒的长度在每次细胞分裂后,被能合成端粒的特殊性DNA 聚合酶-端粒酶所保留。
hnRNPA2 蛋白可与端粒DNA 和端粒酶发生作用,主动打开端粒G-四链体接头,将端粒3` 短的5 个碱基暴露出来,促进其和端粒酶的RNA 模板配对,从而增强端粒酶的催化活性和进行性,可以调控端粒长度平衡,维持细胞分裂和增殖能力。本研究中细胞的永生化诱导方案即通过端粒酶逆转录酶过表达实现。为进一步验证细胞诱导效果,对端粒酶和A2 蛋白进行免疫荧光标记,结果显示(图6):在永生化诱导的细胞中hnRNP A2 蛋白的表达与端粒酶活性呈正相关,两个蛋白共定位于卡佳尔体和端粒,hnRAP A2 的表达促进了端粒的延长。此外,在成体细胞中,端粒酶是无法被检测到的,而在本诱导细胞株中,可以检测到端粒酶的表达,证明细胞的诱导成效显著。另外,hn-RNP A2 蛋白的表达促进端粒DNA 的延长,进一步说明细胞端粒长度缩短机制被打破,端粒开始延长或者延缓缩短,获得细胞永生化诱导细胞株。

图6 hnRNPa2 及TERF2 免疫荧光染色Fig.6 hnRNPa2 and TERF2 immunofluorescence staining
2.7 端粒酶逆转录酶及hnRAP 蛋白表达活性鉴定
端粒作为短的多重复非转录序列,与细胞老化和机体衰老存在最直接的关系,相较于端粒的长短,端粒变短的速度具有更重要的意义。本研究中通过定量PCR 对永生化诱导细胞株(传代180代)和常规细胞株(传代30 代)对比,检测端粒长度和变短的速率,以期验证永生化细胞诱导效率。结果表明:在正常培养状态下,细胞无法传代至180 代,而对照组中的细胞为30 代细胞,对比结果显示:永生化诱导的细胞相对端粒酶长度显著高于对照组(图7a),从侧面揭示细胞永生化诱导的效率高,细胞传代能力和细胞活力显著高于对照组。此外,hnRAPC 蛋白活化对端粒酶活性具有激活作用。本研究中对hnRAPC 基因表达量进行定量PCR(图7b),结果显示永生化诱导组的表达量显著高于对照组,揭示该蛋白在端粒中的活化功能,以及诱导组细胞蛋白活性显著高于对照组,表明永生化细胞诱导获得成功,细胞传代能力得到显著提升。

图7 端粒酶逆转录酶及四聚体表达量分析Fig.7 Analysis of expression of telomerase reverse transcriptase and tetramer
3 讨论
细胞培育肉作为一门新兴科学技术,虽然研究时间相对较短,但是其作为细胞生物学、组织工程学及食品科学等多学科交叉融合的技术已经具备深厚的研究基础[23],尤其是细胞培育肉种子细胞的使用方面,可供细胞培育肉种子细胞使用的细胞类型主要有胚胎干细胞、诱导多能干细胞、间充质干细胞等多能干细胞以及包括肌卫星干细胞、脂肪前体细胞等单能干细胞[24-26]。值得注意的是多能干细胞的优势在于其具有干细胞特性,可以无限增殖和定向分化,而多能干细胞在增殖过程中的干性维持尚未完全阐明,在细胞定向分化过程中,存在批量性和统一性的问题,容易产生不定向分化,同时其培养成本相对较高,不适合于细胞培育肉的规模化制造[27];而单能干细胞在使用过程中,一旦活化,细胞便发生不可逆的分化过程,并且随着干性的消失,细胞传代次数受到限制,制约着细胞的规模化增殖[28]。因此,在细胞培育肉研发过程中,获得相应的可以无限增殖的成体细胞,是解决其工业化过程中细胞来源的重要保障。
在基因水平层面,细胞永生化的方法目前主要有SV40 大T 抗原法[29]、端粒酶逆转录酶法[30]、HPV16 E6 法[31]、原癌基因以及抑癌基因[32]等方法。SV40 主要通过SV40 抗原片段整合到靶细胞的细胞核中并表达,从而构建永生化细胞系。端粒酶逆转录酶法则是基于端粒酶蛋白部分的催化亚基编码基因调控端粒酶的活性,进而诱导细胞永生化。HPV16 E6 是通过病毒编码蛋白与宿主细胞的调节蛋白形成复合物,破坏细胞有丝分裂和DNA 修复过程,进而激活端粒酶而实现正常细胞的永生化。诸如c-Myc、RAS、c-fos、Bmi1 及CDK4等原癌基因可以激活端粒酶的活性,进而促进细胞向永生化过渡;相反的,p53、p21、pRb 等抑癌基因则是通过基因的干扰或者敲除,从而抑制细胞老化,反向协同端粒酶的激活,实现细胞的永生化。这些细胞永生化的方法各有利弊,作为食品科学研究,细胞培育肉使用的种子细胞首先需要排除人源因素,同时保证细胞的活性及各项功能,更重要的是细胞永生化的诱导不能干扰各类种子细胞的正常使用,即不能引入不确定因素。
本研究通过端粒酶逆转录酶载体法诱导细胞永生化,端粒酶逆转录酶导入细胞后随着细胞分裂过程,同步表达逆转录酶的催化亚基,调节端粒酶的活性[33]。同时与hnRNPA2 蛋白发生作用,打开端粒G-四链体结构,暴露端粒3` 端碱基,促进其与端粒酶RNA 模板配对,从而增强端粒酶的催化活性及进行性,有效维持端粒酶的长度,其意义在于减低(负调控)端粒缩短的速度,使端粒维持一定的长度,从而实现细胞的永生化[34]。本研究中永生化细胞的特点是:除了端粒酶逆转录酶,不再引入新的影响基因,以保证细胞的正常表达水平和功能。细胞在传代180 代后仍具有较高的增殖和分化效率。通过定量PCR、免疫荧光标记等手段验证诱导细胞株结果,可以推测细胞稳定性较高,没有发生不可预测的突变现象。综上所述,建立了一株可以稳定表达、高效传代的猪成肌细胞永生化细胞系。
4 结论
本研究构建猪成肌细胞永生化诱导载体,探索细胞永生化诱导方案和研究材料;通过细胞核转染、阳性细胞流式细胞仪筛选获取阳性细胞株;通过定量PCR、免疫荧光标记、核型分析等验证细胞的稳定性;对阳性细胞进行连续传代培养,验证细胞可传代超过200 代。本研究的实施为细胞培育肉种子细胞的开发探索了理论基础,提供了工业化生产的细胞材料,为培育肉工业化生产提供先导条件。
猜你喜欢
中国乳品工业(2022年4期)2022-05-19
西南林业大学学报(2021年3期)2021-05-03
医药前沿(2018年22期)2018-01-17
分析化学(2017年12期)2017-12-25
科学与财富(2016年32期)2017-03-04
北方牧业(2016年6期)2016-12-17
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
恋爱婚姻家庭·养生版(2016年5期)2016-05-06
家庭医学(2015年6期)2015-07-03
创新时代(2015年6期)2015-06-29
