
原花色素对鸡胸肉肌原纤维蛋白凝胶特性影响
2023-05-11 13:29郑卓琦李松艳冯冠霖李静媛
中国农业大学学报 2023年5期
郑卓琦 李 岩 李松艳 冯冠霖 李静媛
(青岛农业大学 食品科学与工程学院,山东 青岛 266109)
鸡胸肉作为一种经济实惠的食品,由于鸡肉营养丰富,适宜微生物生长繁殖,极易腐败且腐败问题严重制约中式快餐企业规模化的推进。鸡胸肉质量主要由其肌原纤维蛋白凝胶的质构、流变等特性决定。提取肉制品中的肌原纤维蛋白是对肉制品中蛋白质性质专门研究的一种方案。肌原纤维蛋白凝胶的形成是基于蛋白质的盐析作用,使蛋白质在溶液中发生团聚和沉淀。凝胶的稳定性在一定程度上是蛋白质稳定性的体现[1]。热加工是生产肌原纤维蛋白凝胶的常用方法之一[2]。在蛋白质的加热过程中,蛋白质骨架断裂,疏水基团暴露。随着温度的升高,蛋白质聚集,α-螺旋结构展开,逐渐转变为β-折叠结构。蛋白质的凝胶化来源于蛋白质之间通过共价键和非共价键的交联,包括离子键、氢键、疏水作用、二硫键等共价键[3-4]。通过研究凝胶的保水性、凝胶强度、抗氧化性等因素,可以从侧面反映出蛋白质的一些生理特性。因此,提高鸡胸肉肌原纤维蛋白的保护机制以及其肉制品品质具有重要意义。
原花色素是广泛存在于植物中的一类黄酮类化合物,是天然抗氧化剂。Xu等[5]研究发现,表没食子儿茶素没食子酸(EGCG)的添加可提高大豆分离蛋白的成胶速率,形成更均匀的网络结构,从而改善大豆分离蛋白的凝胶性能。Ferreira等[6]发现原花色素富含疏水芳香环和羟基,可通过氢键和疏水作用与蛋白质相互作用,通过填充作用支撑和稳定蛋白凝胶的网络支架,还能与蛋白质产生相互作用,有效增强凝胶强度和持水能力。Jia等[7]研究发现,当儿茶素添加量大于2.9 g/kg蛋白时,儿茶素引起的疏水结构域被暴露,凝胶中的巯基(S-H)含量明显降低,在超过14.5 g/kg时凝胶严重恶化,在此浓度下儿茶素对凝胶产生了极不利的影响。Lee等[8]研究表明,儿茶素或原花青素的添加对3D打印材料有强化作用,且添加物具有更好的打印性和凝胶性。研究表明,原花青素可以降低凝胶的表面疏水性,从而改变蛋白质的二级结构,添加原花青素可以保护肉制品免受紫外线诱导的光损伤,是减轻紫外线诱导的氧化损伤的有效策略[1,9]。
原花色素现主要应用领域为鱼油微胶囊的壁材和部分保健食品的添加剂,但在研究中发现其潜在的生物功效并没有很好的应用于医疗或其他领域,在很多食品领域的功能尚未开发完全。本研究设计一定浓度范围原花色素对鸡胸肉肌原纤维蛋白的影响,以及添加原花色素后,凝胶理化性质随时间的变化。本研究目的在于通过研究原花色素对蛋白凝胶特性的影响,反应其对鸡胸肉中最重要的蛋白肌原纤维蛋白的影响,从而探究原花色素是否可以作为一种天然的食品添加剂加入至鸡胸肉中。Cao等[10]研究了添加0.1 g/kg原花色素单倍体可以提高凝胶的氧化稳定性,但是Jongberg等[11]研究发现在添加量为0.5 g/kg时,原花色素对肉及其凝胶产生了负面影响,其基质包括触觉、味觉和视觉与空白组对比均出现明显差异,且凝胶形成不稳定。本研究补足了0.1~0.5 g/kg浓度区间原花色素对凝胶的影响,研究了原花色素对凝胶影响的关键浓度,确定了原花色素的最适添加量及在合适添加量的条件下凝胶的变化情况,对原花色素的实际生产应用提供了可参考的应用价值。
1 材料与方法
1.1 试验材料
新鲜鸡胸肉购自大润发超市。儿茶素、原花色素二聚体和原花色素四聚体购自国药集团化学试剂有限公司,纯度为99.9%。葡萄籽购自山东烟台部分酒庄。
药品EDTA、EDTA-2Na、NaCl、Tris-HCl、Triton X-100、Na2HPO4、NaH2PO4、香兰素和浓硫酸为分析纯,均购自国药集团化学试剂有限公司。药品CH3CN、HOAc、CH3OH、C3H6O、儿茶素为色谱纯,购自国药集团化学试剂有限公司。
1.2 试验方法
1.2.1 原花色素制备与检测
参考李春阳[12]和周蒙等[13]、杨成东[14]和张兵兵等[15]的试验方案,称取20 g葡萄籽用冷冻-研磨机研磨,研磨后的粉末过100目筛。过滤后的粉末用索氏抽提器抽提8 h去除油脂和脂肪。之后用真空冷冻干燥机对溶液进行冻干。冻干后将冻干粉取出,按料液比1∶14加入50%乙醇中,再加入0.56 g/L纤维素酶,静置30 min。将溶液微波消解,微波处理时间6 min,微波功率800 W,微波温度60 ℃。后将溶液在4 ℃下8 000 g离心10 min,取上清液冻干,得到的冻干粉即为原花色素粉末。
参考石磊等[16]的方法,采用香草醛-盐酸法检测原花色素的浓度。根据周蒙等[13]和樊金玲[17]的试验方法,采用高效液相色谱-荧光检测器(HPLC-FLD)检测提取的原花色素组分。使用C-18液相色谱柱,流速为1.0 mL/min,柱温35 ℃,以0.1%磷酸水溶液为流动相A,甲醇为流动相B, 进行梯度洗脱。
1.2.2 凝胶的制备
参考鲁小川等[18]的方法并加以修改, 将切碎的鸡胸肉进行多次盐析去杂并通过三层纱布过滤去除结缔组织和脂质,在10 000 g离心10 min后,用冷蒸馏水洗涤沉淀2次。本研究中使用的蛋白质是从最终收集的沉积物中获得的,将提取的肌原纤维蛋白粗粉(Myofibrillar protein crude powder, MP)粉末与磷酸盐缓冲液混合,形成浓度为100 mg/mL的MP溶液。将50 mL MP溶液置于80 mL烧杯中。此时,分别向烧杯中添加0.05、0.10、0.15、0.20、0.25和0.30 g/kg 原花色素。并将其置于水浴中以1 ℃/min的速度从室温升至80 ℃,在黑暗条件中进行,80 ℃保温30 min。然后将烧杯转移至-20 ℃冷却10 min,弃去液体,剩余部分为合成的凝胶。
1.2.3 凝胶色泽测定
根据Filipecki等[19]的方法对凝胶色泽进行测定,采用全自动色差仪进行检测,检测样品均被切成小立方体(1 cm×1 cm×1 cm)上机检测。
1.2.4 凝胶微观结构
凝胶微观结构采用拉曼显微镜进行观测并记录,样品被切成1 mm薄片进行检测。
1.2.5 凝胶结构强度的测定
根据王秀娟等[20]研究的方法,制备的MP凝胶经过全自动切割机切成小立方体(1 cm×1 cm×1 cm),采用P50探针测试硬度、咀嚼性和弹性,并根据质构剖面分析(TPA)计算出最佳测试条件为:应变(40%)、触发力(10 g)、测试前、测试中和测试后速度(1、0.5和1 mm/s)和间隔时间(10 s)。
1.2.6 傅里叶变换红外光谱和拉曼光谱光谱分析
参考林玲[21]的研究方法,取小部分MP凝胶在真空冷冻干燥机中冻干后研磨成粉,采用KBr压片法制备样品上机检测。扫描波长为500~4 000 cm-1,分辨率为4 cm-1,扫描频率为64。
根据薛思雯[22]研究的方法并根据仪器调试参数,激光功率10.0 mW, 曝光时间2 Hz, 扫描次数350。
1.2.7 低场核磁共振
根据孙任宽等[23]的研究方法并做了优化,谐振频率为22.6 MHz、累加次数NS为32,在室温条件下对水的迁移率和分布进行了评估。将约2 g MP凝胶置于直径为15 mm圆柱形玻璃管中,插入核磁共振探针中检测。
1.2.8 流变学特性
根据汤回花等[24]的研究方法,采用配备钢平行板几何结构的流变仪评价黏弹性性能。将未加热的1.5 mL MP溶液置于平板表面,立即用硅油密封。采用10 Hz的恒定频率,在以下程序下监测储能模量(G’)和损耗模量(G”):以0.5 ℃/min从30 ℃加热至80 ℃。此外,对热处理结束(80 ℃)的凝胶样品进行了频率扫描测试,应变范围为0~100 Hz。
1.2.9 硅压阻式力敏传感器测量凝胶体积
根据陈莹梅等[25]的研究方法并改进,将制备好的凝胶用超声波切割机切割出1 cm×1 cm×1 cm的立方体。将切下的样品放入检测器中10 s后读取,可以防止因凝胶表面张力而造成的数据误差。
1.3 统计分析
每份样品测定3次。试验采用origin软件绘制图表和IBM SPSS Statistics软件分析数据显著性差异和误差等。
2 结果与分析
2.1 原花色素提取分析
以儿茶素标准溶液浓度为横坐标,吸光度为纵坐标,绘制儿茶素标准曲线。回归方程:Y=0.001 191X+0.001 23,R2=0.999。根据公式最终提取率为(4.69±0.11)%(w/w),相对偏差为0.638%。
根据图1液相色谱图数据分析,该方法提取的原花色素主要成分为原花色素二聚体,其中儿茶素、原花色素二聚体和原花色素四聚体的比例约为5∶31∶4,因为原花色素二聚体相较于儿茶素具有更高的抗氧化活性[13],故提取的原花色素对凝胶抗氧化作用应存在有利影响。由图1可知,从葡萄籽中提取的原花色素含有儿茶素、二聚体和四聚体。但出现连峰和杂峰的情况,是因为原花色素提取物中所含成分比较复杂,有许多的同分异构体[26]。
2.2 凝胶特性分析
图2显示的是添加不同浓度提取的原花色素后形成的凝胶外观。从图2和表1色差仪检测的数据可以看出,由于提取的原花色素呈现鲜红色,故添加后对所产生的凝胶色泽有所影响,且当原花色素添加量≥0.30 g/kg时,凝胶的状态不稳定且外观架构及颜色中的白值不稳定,没有研究价值,最终确定为0~0.20 g/kg原花色素添加量。
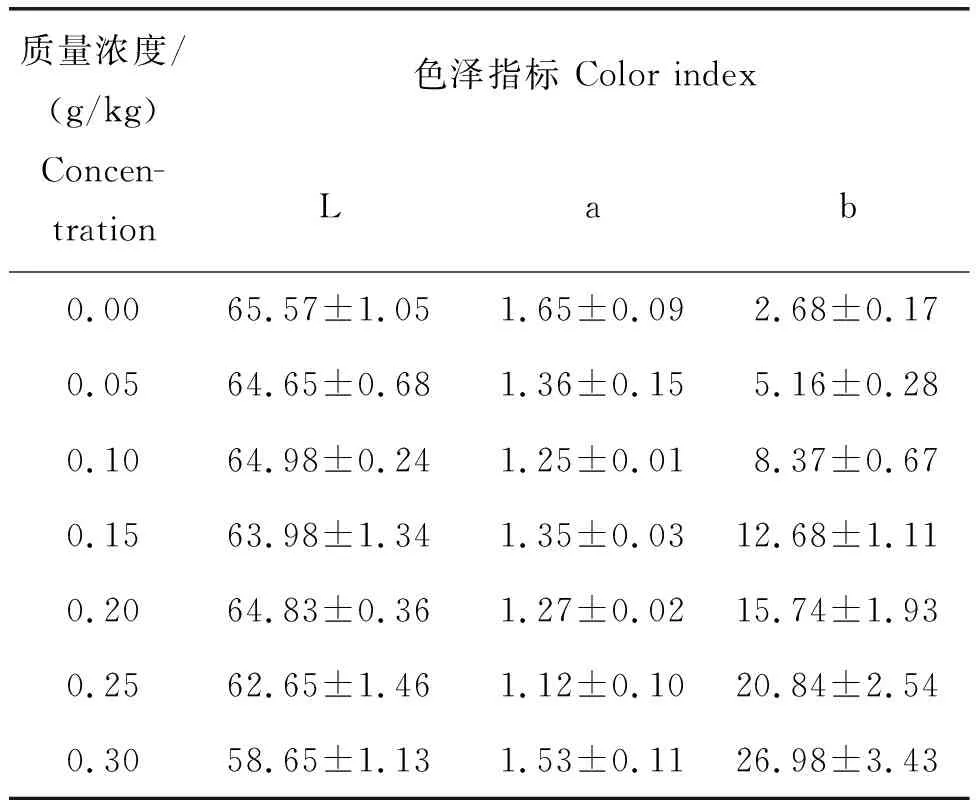
表1 不同浓度原花色素对颜色的影响Table 1 Effects of PAS on gel chromatic aberration
采用双缩脲法对提取的蛋白质进行检测,测得纯度为(90.08±2.27)%,与先前研究[27-28]开展凝胶研究的蛋白质提取纯度大致相同。
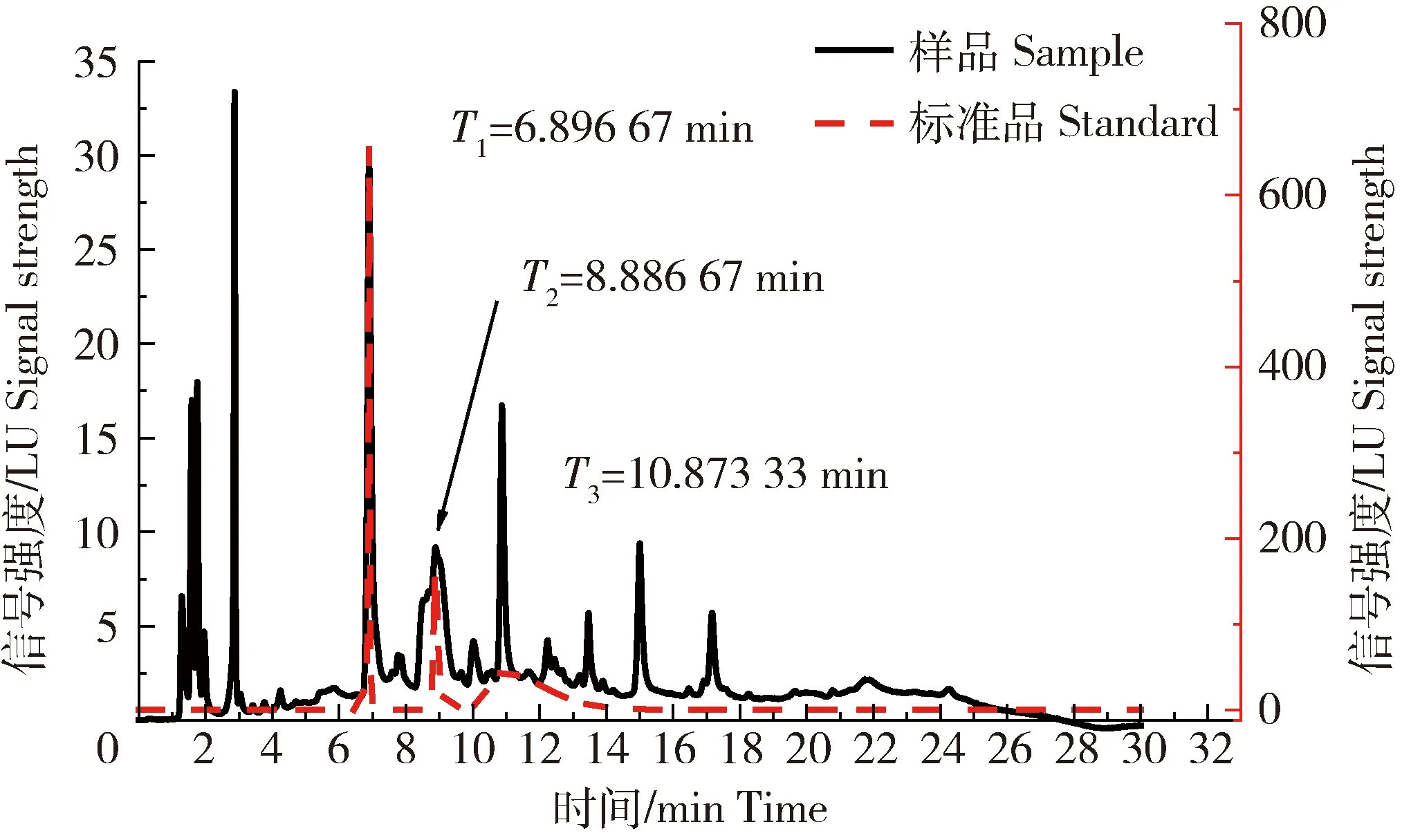
红色曲线为含儿茶素、原花色素二聚体和原花色素四聚体的混合标品的HPLC-FLD色谱图,黑色曲线为冻干法后重新溶解的原花色素的HPLC-FLD色谱图,图中T1-3为物质出锋时间。The red curve was the HPLC-FLD of the mixed standard substance containing catechin, proanthocyanin dimer and proanthocyanin tetra. The black curve was the HPLC-FLD of the proanthocyanin redissolved after lyophilization. T1-3 in the figure was the material retention time.图1 提取的PAS与标品的HPLC-FID高效液相色谱图Fig.1 Label and extracted PAS of HPLC-FID
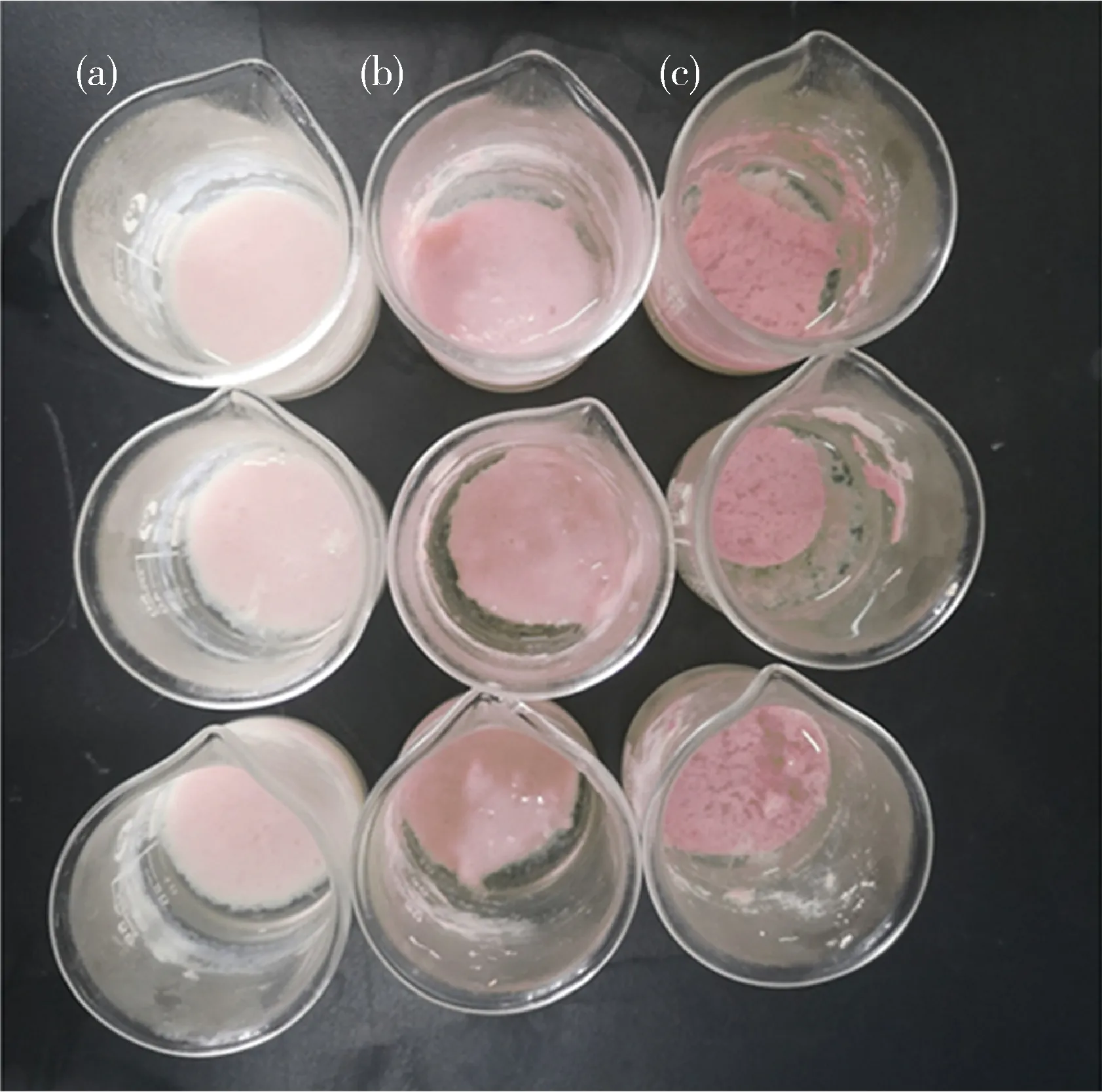
(a)、(b)和(c)列分别为添加了0.10、0.20和0.30 g/kg原花色素后形成的凝胶形态。Column (a), (b) and (c) showed the gel after adding 0.10, 0.20 and 0.30 g/kg PAS.图2 不同浓度原花色素对凝胶外观的影响Fig.2 Effects of PAS on gel appearance
2.3 凝胶表征分析
2.3.1 凝胶含水量及体积分析
保水性是评价肉及肉制品品质的重要指标之一,在加热过程中蛋白结构的展开、变性和聚集以及与原花色素的聚合作用之间的相互作用都会影响凝胶网状结构的形成,并最终改变凝胶的保水性[29]。图3中3个峰从左到右依次为结合水、不易流动水和自由水,当H原子处于不同分子上或处于不同物理状态下的样品中时,其弛豫时间会有所不同[30]。从图4可以看出,在添加原花色素的凝胶形成的0 d,3种水的弛豫时间对比空白组均显著延迟,说明形成的凝胶中的H原子核能被激发后恢复到原来平衡状态所需的时间增多,H原子核可以储存更多的能量,凝胶系统由一个平衡态到达另一个平衡态所用的平均时间间隔更长,说明了原花色素的加入使得凝胶中H原子核需要吸收更多的能量才会发生改变,侧面说明了原花色素增强了凝胶持水的稳定性。弛豫时间的延迟证明原花色素对凝胶的水分子的固定起到了增强作用。其原因可能是部分凝胶分子在聚合过程中与原花色素形成了具有较多结合水的空间结构,也可能是原花色素在凝胶形成阶段与大量羟基反应使H原子暴露较多。
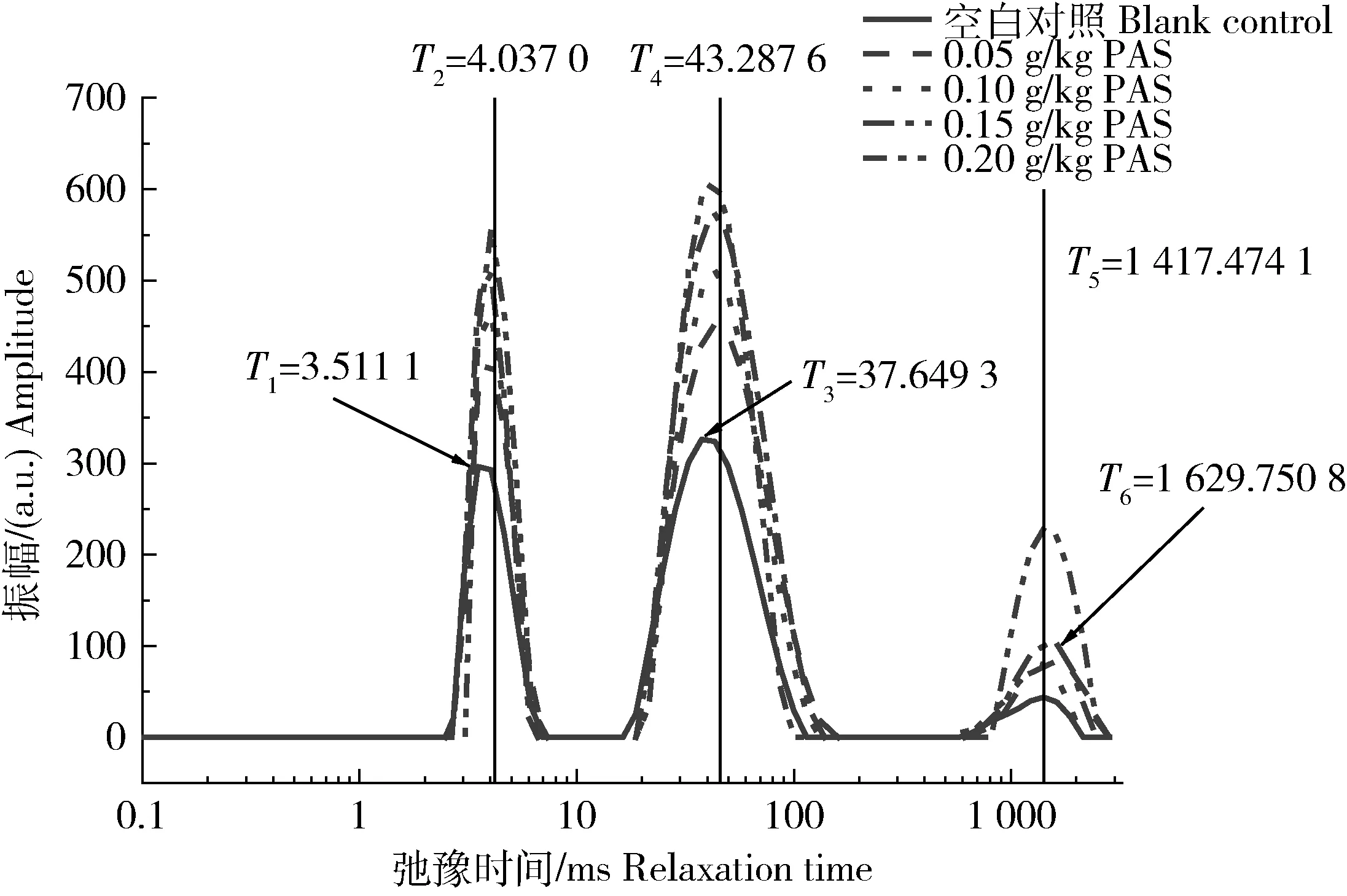
T1、T3、T5为空白组的结合水、不易流动水和自由水的出峰时间;T2、T4、T6为添加原花色素组的结合水、不易流动水和自由水的出峰时间。T1, T3 and T5 were the peak time of combined water, immobilized water and free water in the blank contrast; And T2, T4 and T6 were the peak time of combined water, immobilized water and free water in the PAS group.图3 凝胶形成时低场核磁共振图Fig.3 Low-field NMR during gel formation
图4和表2给出了5个样品随时间变化的低场核磁共振检测图。在随时间变化的检测中,只有T1和T2的弛豫时间在5 d及以后的检测中再次提前。其原因可能是原花色素的羟基与凝胶的部分氨基酸分子在空气氧化作用下形成了较为稳定的结合水。也就是说,原花色素诱导了游离水向固定化水的转化,凝胶中的水分流动受到了某种限制,这种现象可能归因于良好的聚集性。
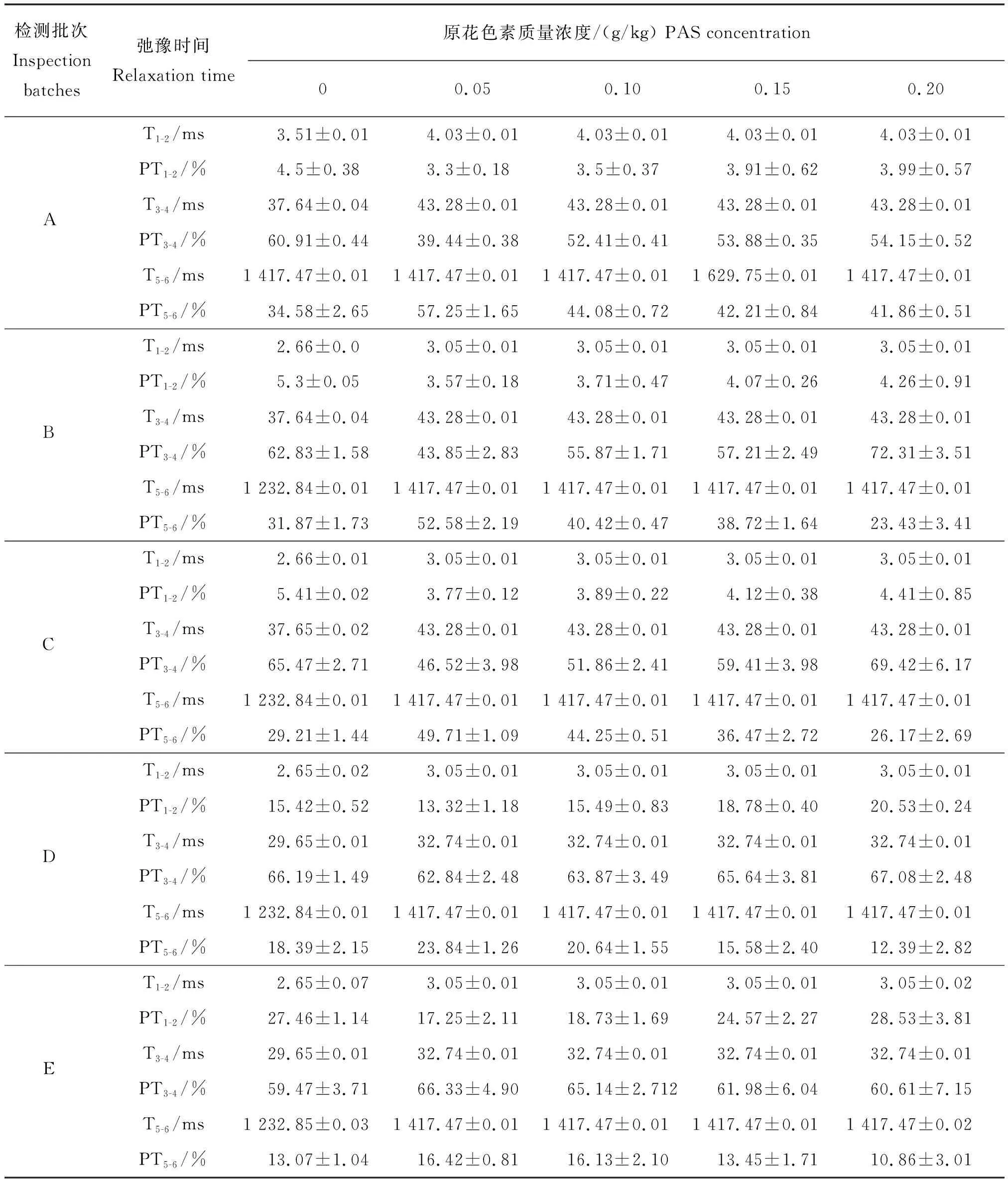
表2 凝胶形成后随时间推移的弛豫时间及占比Table 2 Relaxation time and proportion with gel formation over time
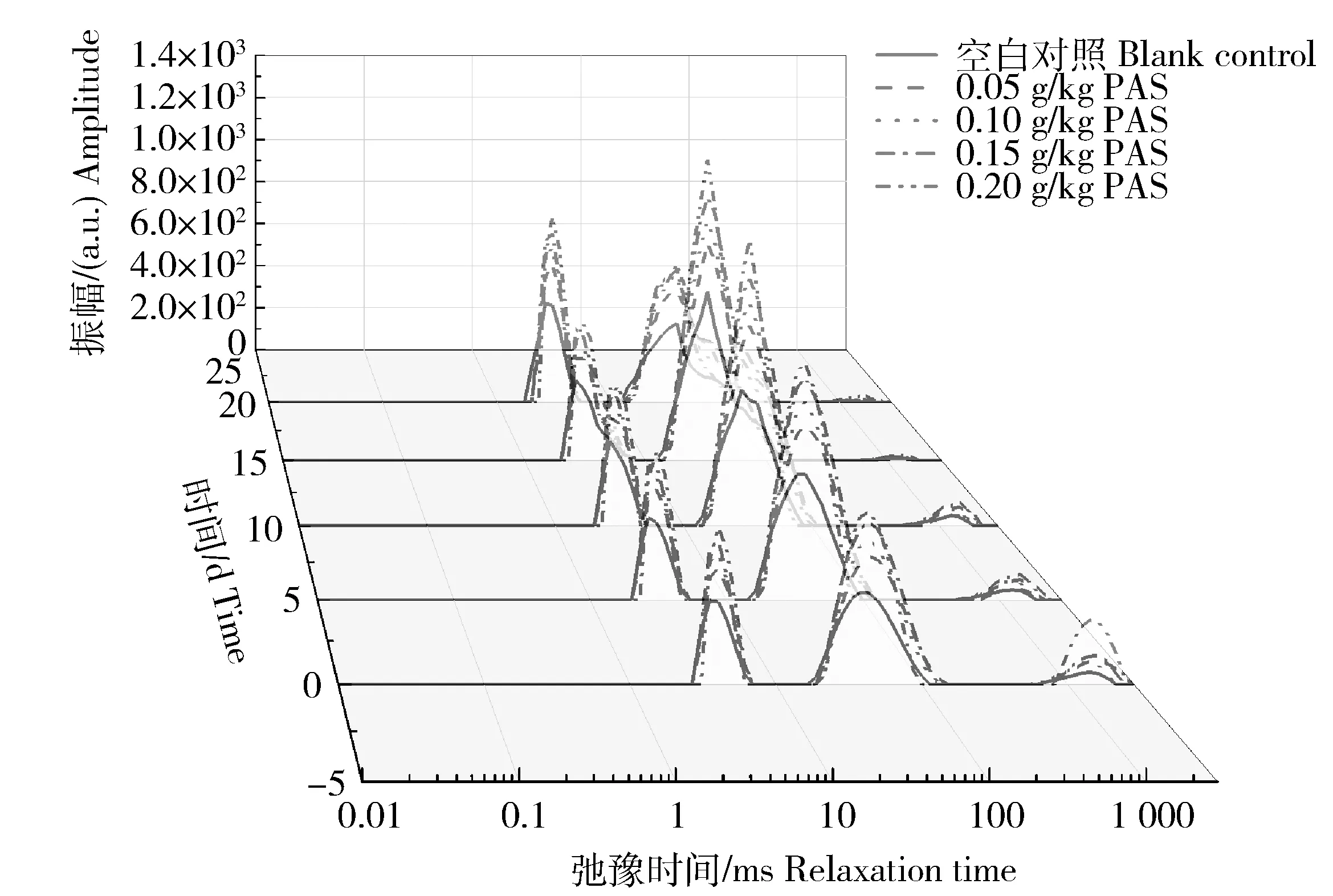
图4 凝胶形成后随时间推移的低场核磁共振图Fig.4 Low-field NMR change gel formation over time
从图5可以看出,在0 d时,虽然切割参数设定为1 cm×1 cm×1 cm,但硅压阻式力敏传感器测得的体积仍不足1 cm3,这可能是因为切割机的误差,也可能是凝胶形成时结构不均匀或产生大孔导致。由数据可知,原花色素加入后形成的凝胶具有更紧密的空间结构,凝胶密度也更大,水分子不易挥发,保水性更强。前10 d时,凝胶体积与原花色素添加量呈正相关。10 d后,添加0.15和0.20 g/kg原花色素的凝胶体积迅速下降,但仍高于空白对照,可能是由于凝胶表面氧化孔隙变大变软导致测量时散落。因此,可以推断,在添加0~0.2 g/kg原花色素浓度下,凝胶的保水性随着时间的推移相比于空白组更加稳定,高浓度原花色素短期保水性较好,但长期保水性下降较快。
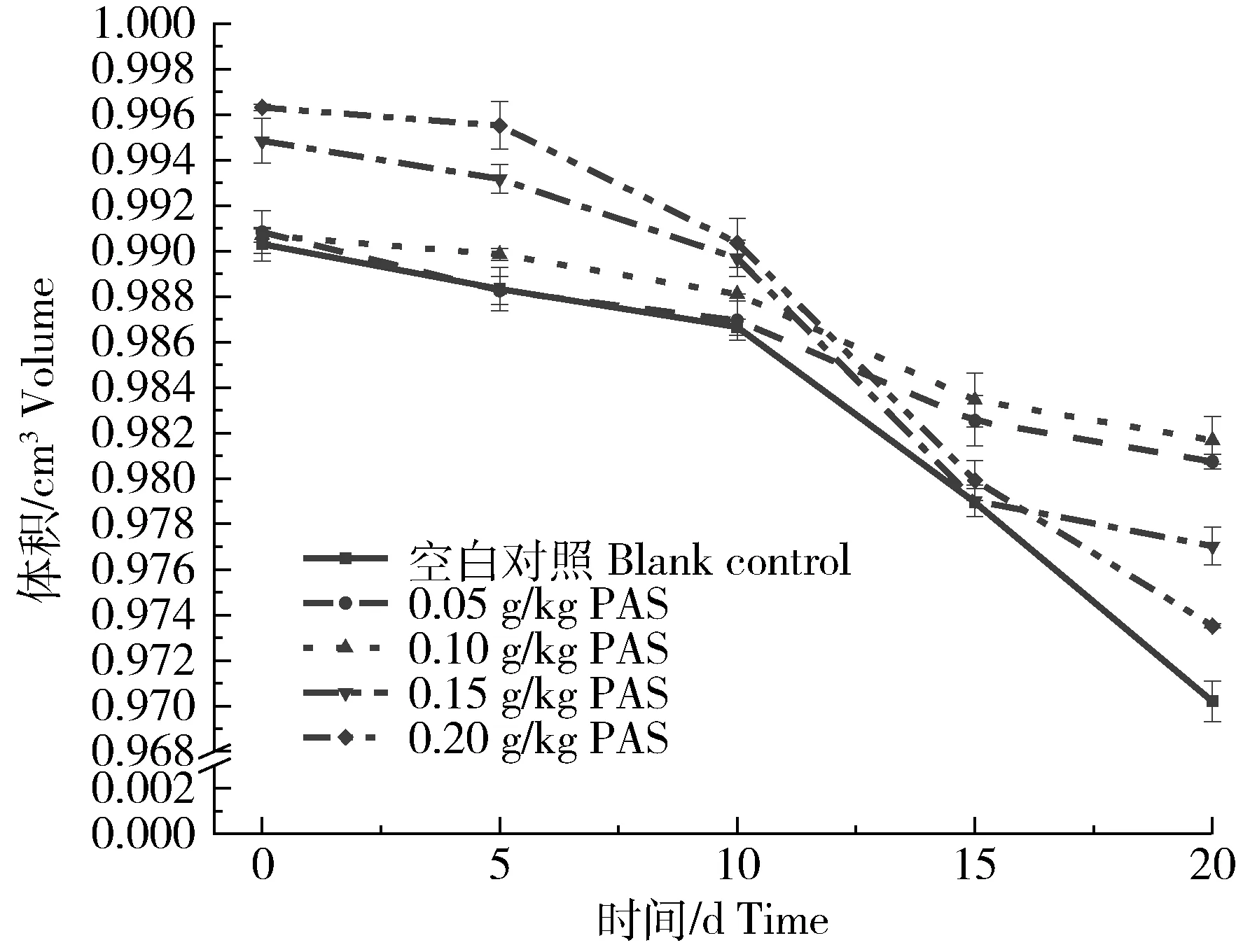
图5 凝胶随时间推移的体积变化Fig.5 Volume change of gel over time
2.3.2 红外光谱和拉曼光谱分析
利用红外光谱进一步研究了蛋白质-原花色素分子间的相互作用。如图6所示,中心位于3 000~3 600 cm-1的宽带归属为O-H和N-H基团,暗示了氢键相互作用的潜力增强[31-32]。这意味着随着原花色素的加入,凝胶力中O-H和N-H基团增加,也可能是原花色素本身自带的O-H和N-H基团导致了氢键相互作用的增加。与空白相比,O-H和N-H基团在3 000~3 500 cm-1处出现伸缩振动,0.15%和0.20%原花色素试验组的强度增强,表明分子间和分子内氢键强度增强。这是影响蛋白质之间亲和力的有利变化。MP凝胶表面的强氢键势有助于蛋白质之间的稳定和有序,加速了蛋白质的重排和聚集过程[9]。在2 300 cm-1左右是产生气体的峰,这与凝胶生产时观察到的表面一致,随着原花色素浓度的升高,凝胶表面出现的微气泡增多且更加透软,但切开后内部仍十分紧实,这可能是蛋白质的某些集团与原花色素活跃的羟基发生反应产生的气体有关。

图6 凝胶形成时的红外光谱图Fig.6 Infrared spectrum of gel formation during gel formation
近年来,许多研究利用拉曼光谱来反映蛋白质凝胶的二级和三级结构信息。凝胶性质与二级结构的变化有关。通常用酰胺Ⅰ(1 600~1 700 cm-1)和酰胺Ⅲ(1 200~1 300 cm-1)的拉曼光谱来测量二级结构[19]。图6显示了0 d时不同浓度原花色素处理后凝胶的拉曼光谱。通过高斯曲线和傅里叶自反卷积拟合分析以1 652 cm-1为中心的酰胺Ⅰ带和以 1 234 cm-1为中心的酰胺Ⅱ带和波长为1 509~1 592 cm-1为酰胺Ⅱ区,分析蛋白的结构。1 652、1 663、1 674和 1 683 cm-1的条带分别为α螺旋、无规卷曲、β-折叠和β转角[33]。(1 235±10) cm-1波长是β-折叠在酰胺Ⅲ带的表现[34]。920~1 180 cm-1波长为N-Cα-C骨架伸缩振动,被一些作者归类为酰胺模式[35]。
由图7可知,添加原花色素的凝胶中,α-螺旋总量与原花色素添加量无明显相关性,但1 650 cm-1左右酰胺Ⅰ带对比空白组的拟合峰面积都减少了,这意味着α-螺旋总量的减少。这种减少可能是由于凝胶主要部分中相当数量的α-螺旋的解旋化造成的。需要指出的是,α-螺旋中的肽键可能形成氢键[36],因此,α-螺旋非常稳定。本研究中,加入原花色素后,α-螺旋有很大程度的解旋化,分子内疏水基团的暴露似乎有利于解旋化作用后凝胶网络的形成和新蛋白折叠的出现(β-折叠)。从而促进了疏水作用和凝胶化的稳定。
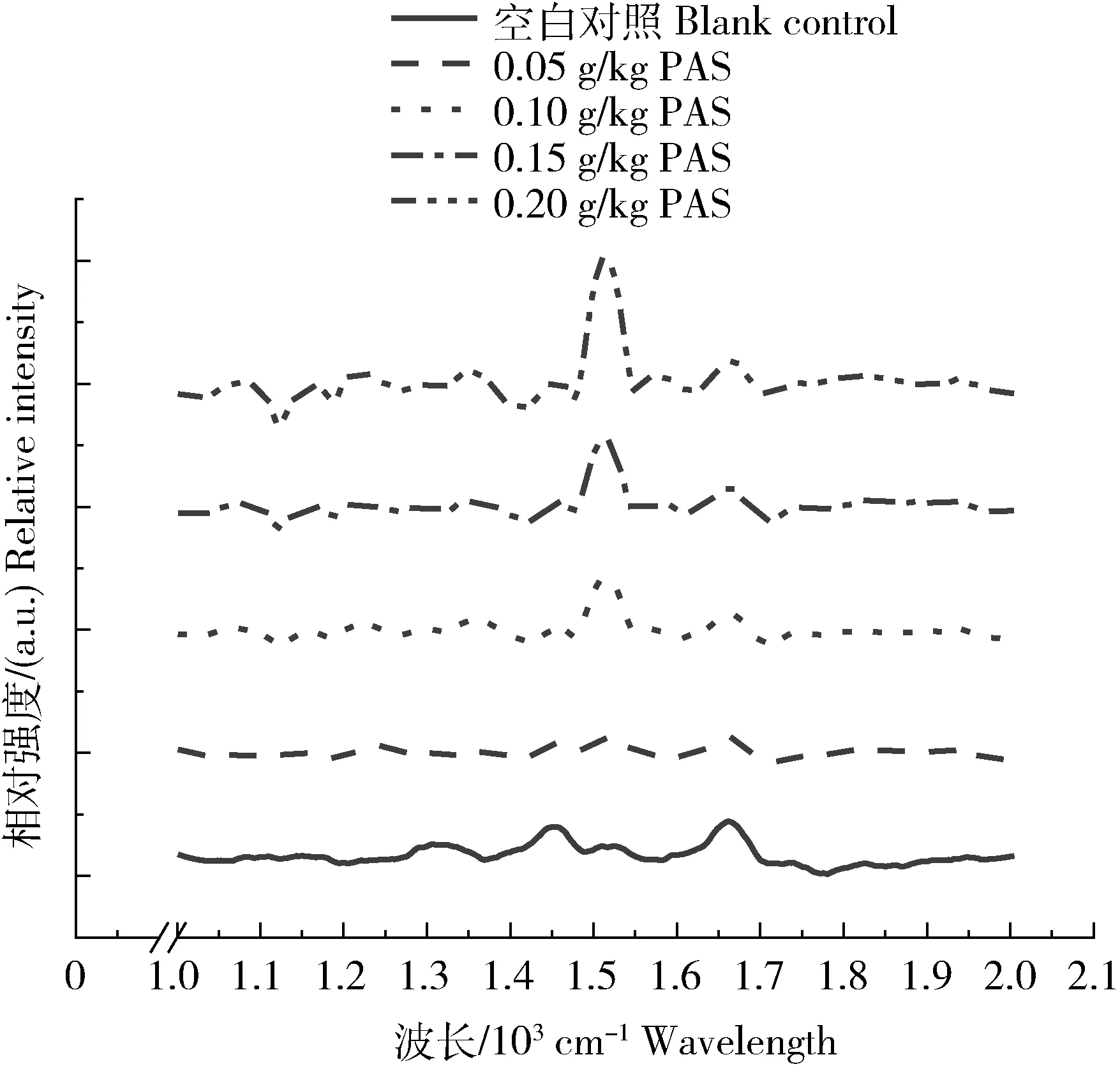
图7 凝胶形成时的拉曼光谱图Fig.7 Raman spectra of gel formation during gel formation
图8(a)和(b)检测到的是酰胺Ⅰ带和酰胺Ⅲ带的β-折叠。在0 d时形成凝胶时,添加了原花色素形成的凝胶中β-折叠的含量均高于空白对照组,这与α-螺旋总量减少,减少的α-螺旋部分转化成了β-折叠推论相吻合。随着时间推移,空白组与试验组的β-折叠含量虽均出现下降趋势,但试验组的β-折叠含量仍高于空白组。这可能是因为添加了原花色素的凝胶在形成过程中原花色素的聚合带走了凝胶中部分氨基酸的电荷。随着相同电荷的减少和不同电荷的增加,肽链之间形成的β-折叠变得更强和更稳定。但随着时间的推移,特别是5~10 d,β-折叠有快速下降的趋势,10 d后继续缓慢下降,可能是凝胶中部分氢键或S-S键被氧化断裂,导致凝胶不稳定。
根据图8(c)可以得出,在凝胶形成的0 d时,原花色素的加入量与N-Cα-C骨架伸缩振动的强度范围呈负相关,这很可能是由于原花色素中大量的C=O、羟基或苯环增强了凝胶中某些氨基酸的N-Cα-C稳定性,抑制了N-Cα-C骨架伸缩振动的振幅,导致检测到的N-Cα-C骨架伸缩振动低于空白组。但随着时间的推移,原花色素的强化稳定作用逐渐减弱,空白组与试验组能检测到的N-Cα-C骨架伸缩振动强度均呈现上升趋势,这可能与凝胶被氧化导致的密度变小、表面硬度降低、凝胶空隙变大有关。
酰胺Ⅱ区有N-H面内弯曲、C-N拉伸、C-O面内弯曲和C-C拉伸[37-38]。根据图8(d)可以得出,随着原花色素的加入,酰胺Ⅱ区的检测强度也增强,可能是由于原花色素分子中存在C-C、C-O键使得检测信号增强。但酰胺Ⅱ区的强度仍随时间的延长而增大。可能的原因是氧化和脱水暴露了酰胺Ⅱ区域的C-C拉伸和C-O面内弯曲,从而检测到了较大的信号强度。
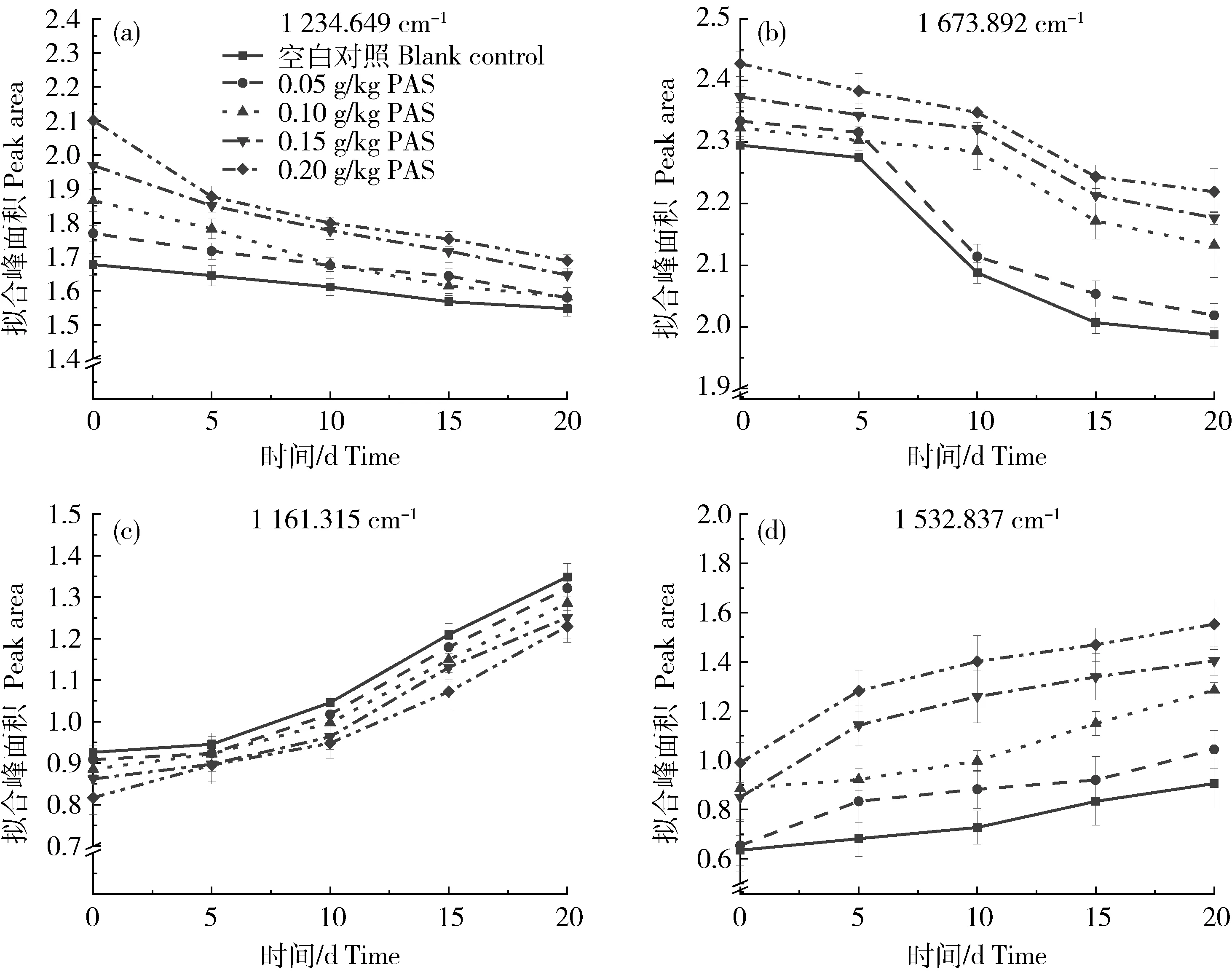
图8 凝胶随时间推移的拉曼光谱部分波长峰面积拟合图Fig.8 Fitting diagram of partial wavelength peak area of Raman spectra
2.3.3 流变性能分析
储能模量(G’)和损耗模量(G”)是表征凝胶粘弹性的重要指标。动态流变分析对研究肌肉加工过程中蛋白质功能特性非常有用,有助于研究凝胶形成过程[29]。从图9(a)和(g)可以看出,随着温度的升高,G’逐渐减小,在48 ℃时达到最低点,然后继续上升到80 ℃,G’继续增加到稳定状态, Sovrani等[39]也发现了类似的结果。G’的增加表明凝胶或弹性蛋白网络结构的初步形成。48 ℃前G’的暂时性下降是由于肌球蛋白重链的伸缩和交联,可能是由于肌球蛋白尾部变性,主要涉及非共价键的解离和分子间的暂时性相互作用,从而增加了肌球蛋白的流动性[8]。进一步升温后,疏水基团的形成和二硫键的相互作用增加了蛋白质聚集体的相互作用,形成了良好的丝状结构,从而导致G’增大,说明黏性溶胶状态向弹性凝胶网络结构转变[40-41]。随后G’迅速增加,在72 ℃时增长放缓,说明凝胶网络结构完全形成。原花色素的添加,使得凝胶形成的G’均高于空白对照组,说明凝胶存储弹性变形能量的能力增加。对流变完成后的样品进行观察,与在水浴锅中凝胶的形成表态相似,形貌为絮状凝聚块。
根据图9(b)和图9(f)可以得出,随着原花色素的加入,G”也呈现上升趋势,G’是G”的5倍多。随着温度的升高,在70 ℃左右达到峰值,之后呈下降趋势。当添加量为0~0.20 g/kg时,凝胶的黏度随添加量的增加而增加,且高于空白对比。从图9(e)可以看出,随着原花色素浓度增加,实验组和空白组的MP溶液的复合模量均继续增加,其粘弹性不断增强,向着形成凝胶的方向发展。从图9(g)和(h)可以看出,随着温度、频率和原花色素添加量的不断增加,凝胶的储能模量和损耗模量都在增加,但储能模量的增长速度明显高于损耗模量的增长速度,证明在加热过程中形成的凝胶,在频率扫描过程中也表现出一定程度的凝胶稳定性。
图9(c)、(d)和(h)中的频率扫描表明,在0~0.20 g/kg质量浓度范围内,随着原花色素和扫描频率的增加,除0.15和0.20 g/kg的原花色素实验组,凝胶的G’和G”均呈现上升趋势。但从添加0.15 和0.20 g/kg原花色素的凝胶的数据可以看出凝胶已经表现出一定的不稳定性。当添加量为0.05 g/kg时,PG的复合黏度数据与空白对比无明显差异。当添加量为0.05~0.20 g/kg时,可以看出原花色素的添加增强了凝胶的强度,但在0.20 g/kg时也存在不稳定性。随着原花色素添加量的增加,凝胶的硬度和黏度增加,但PG在0.20 g/kg时存在不稳定性,这与质构、拉曼和红外检测结果一致。
2.3.4 质构分析
图10(a)中的咀嚼性数据显示,0.05 g/kg原花色素添加量的咀嚼性低于空白组。考虑到原花色素浓度过低时形成的凝胶黏附性和黏聚力不足,对二硫键和酰胺键的影响不稳定,导致0.05 g/kg时咀嚼性较低。但随着原花色素用量的增加,可以看出咀嚼性有非常迅速的增长趋势。随着时间的推移,各浓度试验组中的咀嚼性随时间迅速下降。可以看出,原花色素在凝胶形成过程中可以起到抗氧化和稳定作用,但原花色素容易聚合,低聚物形式过多,在参与凝胶形成时会因为不同集团的种类和数量影响凝胶的稳定性[42]。随着时间的推移,不同的分子基团由于氧化而转变为新的分子形态,导致凝胶外的凝胶层发生软化。将20 d凝胶切片后发现,凝胶表面柔软、柔嫩,无凝胶特性,但中心部分仍显示凝胶特性。这与拉曼显微镜的观察趋势一致。
从图10(b)可以看出,凝胶的弹性与原花色素添加量呈正相关,在4 ℃贮藏20 d后,原花色素组的弹性衰减率低于空白组,衰减率比空白组平均低13.574%。根据数据推测,弹性下降与凝胶20 d内游离水流失和二硫键破坏有关。从图6也可以看出,硅压阻式力敏传感器测量的凝胶体积下降也表明凝胶有一定程度的收缩和塌陷。但从数据来看,实验组的凝胶崩解速率比空白对照组慢。这可能是由于在凝胶形成的加热过程中,原花色素中的C-环与凝胶颗粒中的C-C、C-N键发生聚合,加强了凝胶中键之间的相互作用[29]。从图10(c)可以看出,随着试验组原花色素浓度的增加,形成的凝胶硬度增加,并且随着时间的推移,凝胶硬度仍然增加,这与凝胶的失水直接相关。20 d时,两组自由水含量均显著降低,凝胶呈现硬脆特性。
同时发现,在质构仪检测过程中,实验组比空白组更加不稳定,误差更大。分析认为,在凝胶形成的加热过程中,形成了不同的原花色素低聚物或聚合物,不同的聚合物对S-S键的影响不同,使得试验组的稳定性低于空白组。
2.3.5 微观结构分析
通过对比图11(a)和(b)的显微图像,空白组凝胶表面粗糙,结构紊乱,说明凝胶化过程中蛋白质分子没有充分展开,彼此之间没有很好的连接。随着原花色素的加入,凝胶网络变得更加规则和均匀,从图11(c)和(d)中可以看出形成的凝胶孔隙更加致密且并不会随时间变化使得凝胶结构出现较大程度的变化。高度互联、紧密的网络结构可能会表现出更大的抗外应力,通过毛细管效应为包裹水提供更多的空间,从而提高凝胶强度和保水性。此外,基于原花色素的自我聚集效应,团聚的原花色素可以充当填料填充网络结构,且这些填充效应在较高浓度时似乎更加明显。
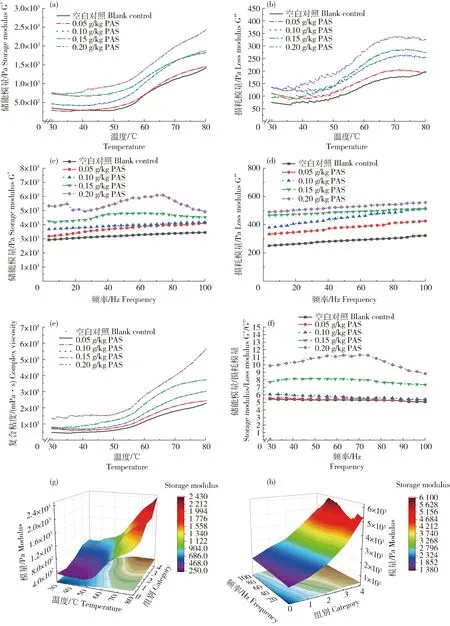
(a)储能模量随温度的变化;(b)损耗模量随温度的变化;(c)储能模量随频率的变化;(d)损耗模量随频率的变化;(e)复合模量随温度的变化;(f) 储能模量/损耗模量G’/G”;(g)储能模量与损耗模量随温度的变化;(h)储能模量和损耗模量随频率的变化。(a) Change of storage modulus with temperature; (b) Change of loss modulus with temperature; (c) Change of storage modulus with frequency; (d) Change of loss modulus with frequency; (e) Change of composite modulus with temperature; (f) G’/G”; (g) Change of storage modulus and loss modulus with temperature; (h) Change of storage modulus and loss modulus with frequency.图9 凝胶随时间变化的流变学数据Fig.9 Rheology data change of gel over time
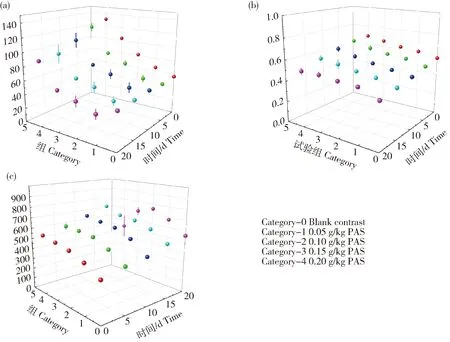
(a)凝胶咀嚼性随时间的变化;(b)凝胶弹性随时间的变化;(c)凝胶硬度随时间的变化。(a) Change of chewness with time; (b) Change of springness with time; (c) Change of hardness with time.图10 凝胶随时间变化的质构数据Fig.10 Texture data change of gel over time
根据图11(a)和(c)可以看出,0 d形成的凝胶表面和内部凝胶结构不同,这与低场核磁共振检测、体积检测和质构检测中发现的问题一致。考虑可能是凝胶形成过程中凝胶外层表皮与溶液过度接触所致。研究发现,如果在0 d时剥除凝胶所有表层部分,那么20 d可见两组凝胶均出现大面积塌陷,考虑凝胶在被氧化的过程中产生了大量的羧基导致。
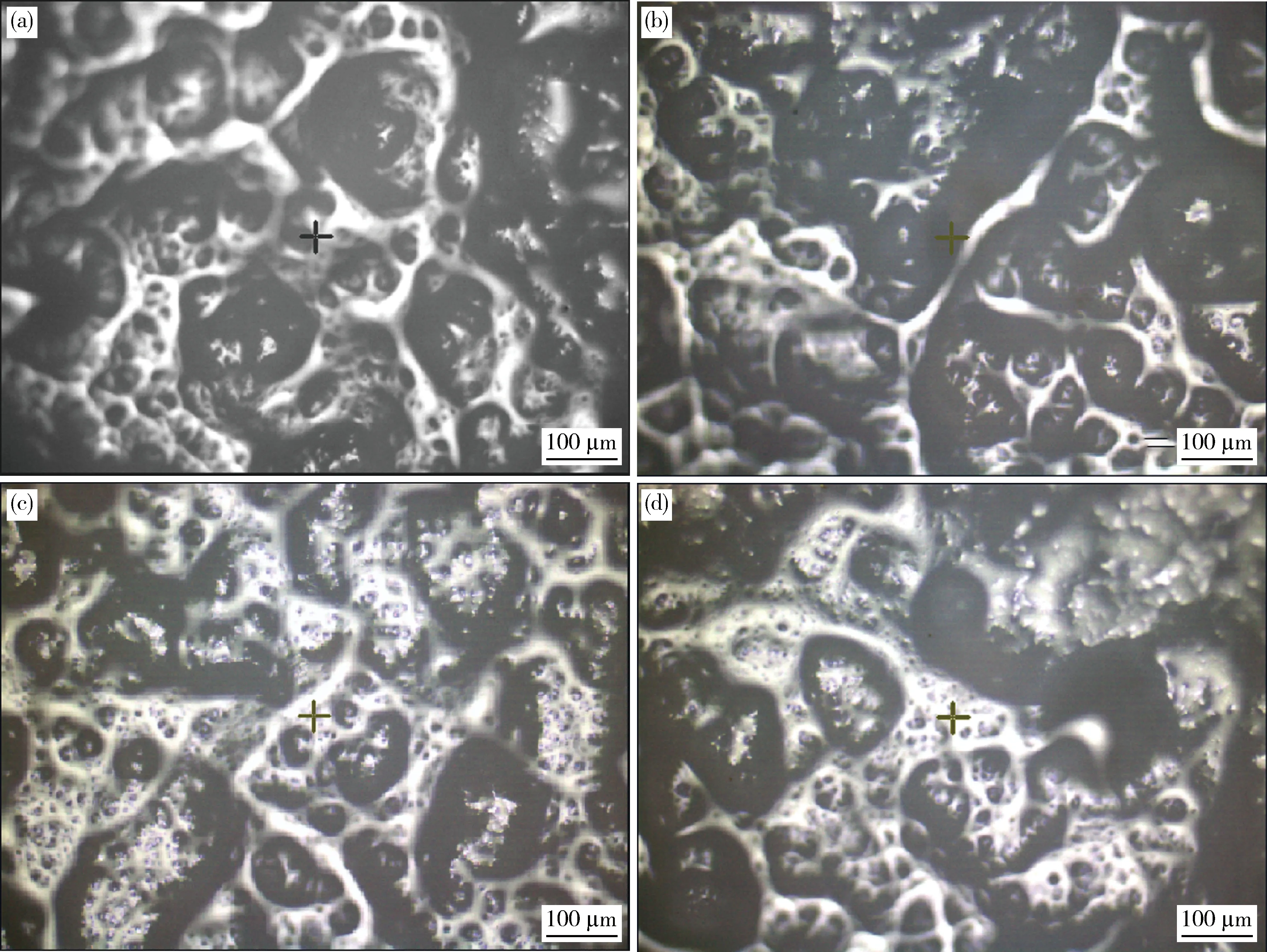
(a)0 d空白对照组;(b)20 d空白对照组;(c)0 d 0.20 g/kg原花色素;(d)20 d 0.20 g/kg原花色素。(a) Blank contrast on day 0; (b) Blank contrast on day 20; (c) PAS group on day 20; (d) PAS group on day 20.图11 凝胶随时间变化的微观图Fig.11 Microscopic graph change of gel over time
3 结 论
本研究填补了Ferreira等[6]与Jia等[7]研究中PAS浓度范围的空缺,找到了PAS浓度对肌原纤维蛋白凝胶影响的拐点。本研究首次引入硅压阻式力敏传感器测量凝胶体积,在原有的凝胶常用表征方法[43-44]上丰富了凝胶数据支撑,使得凝胶表征数据更加完整。通过微观和宏观的结果表明,PAS的加入提升了凝胶保存自由水的能力,减缓了流动水随时间的损耗速度,通过提高凝胶的空间密度,提升了凝胶存储弹性形变能量的能力,这与Xu等[5]研究的大豆分离蛋白凝胶结果相似,说明PAS具有使得凝胶具有更强的保水性以及更致密的空间结构的能力。红外和拉曼结果与Saeki等[45]研究分析的EGCG对凝胶影响趋势相同,PAS通过增加蛋白质二级结构中的氢键之间的强度、促进蛋白质之间的疏水作用、蛋白之间形成更多的β-折叠,从而强化蛋白质的二级结构,使得添加PAS后的凝胶具有更稳定的二级结构和更不易被破坏的空间结构。Lee等[46]研究发现,儿茶素的添加有助于提升凝胶的储能模量,提升水凝胶敷料的愈合性能,加速了凝胶成型的速度,这与本试验中凝胶获得了更高的弹性和更高的硬度以及更加致密的空间结构结论相同,蛋白质之间的作用力更强,凝胶更具有稳定性。凝胶的流变学结果表明,添加PAS后,凝胶更易形成,且在形成凝胶的过程中,储能模量和损耗模量与PAS添加量呈正相关趋势,凝胶具有更强的回弹能力和更大的粘性,这与先前的研究趋势相同[5,46]。因此,PAS作为一种天然且充裕的物质,可作为一种新型的食品添加剂作用于个性化凝胶制品和肉制品研发中。其营养价值的评价与感官评价有待进一步研究。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
陶瓷学报(2021年1期)2021-04-13
军事文摘(2020年20期)2020-11-16
中学生数理化·八年级物理人教版(2020年12期)2020-01-01
中学生数理化·八年级物理人教版(2018年12期)2019-01-31
现代园艺(2017年13期)2018-01-19
衡阳师范学院学报(2016年3期)2016-07-10
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
茶叶通讯(2014年2期)2014-02-27
食品科学(2013年22期)2013-03-11
