
苯甲酸阿格列汀二甲双胍片中阿格列汀相关杂质的控制
2023-05-25 10:01拥青她姆王珍珍丁平田
中国药剂学杂志(网络版) 2023年3期
拥青她姆,王珍珍,丁平田
(1. 沈阳药科大学 药学院,辽宁 本溪 117004;2.江苏豪森药业集团有限公司,江苏 连云港 222000)
苯甲酸阿格列汀二甲双胍片是用于治疗 2 型糖尿病的一种复方制剂,由 Takeda(武田)公司开发,最初于 2013 年 1 月获得 FDA 批准上市(KAZANO)。该复方制剂采用两种不同作用机制的降糖药物联合治疗,可通过“降糖和抑糖”的原理,直击疾病的 3 大缺陷——胰岛素分泌缺陷、胰岛素抵抗和肝糖原过度输出。通过具有互补机制的两种药物联合治疗,能够有效提高患者的治疗效果,延缓疾病的进展,并且还能减少不良反应的发生[1-3]。
复方制剂与单组分制剂相比在技术上的要求更多,尤其是两种活性物质及辅料之间的相互作用及质量控制[4]。目前,苯甲酸阿格列汀二甲双胍片(以下“复方片”)在各国药典中尚未收录,有部分文献分析了阿格列汀原料或片剂中的杂质[5-8],但对该复方片中杂质研究的文献较少。根据苯甲酸阿格列汀的合成路径及可能的降解途径,分析了苯甲酸阿格列汀相关的可能存在的 10 种杂质来源(见图 1),笔者重点对其中 8 种杂质进行了检测方法开发和验证,最终结合参比制剂和自制制剂稳定性结果重点对 5 种已知杂质(杂质 D、E、F、I 和 J)进行质量控制,其中 2 种杂质(杂质 E 和 F)未见文献报道(见表 1)。
1 材料及仪器
1.1 材料
原料药:苯甲酸阿格列汀原料药(江苏豪森药业集团有限公司自制,批号:AL-131001),盐酸二甲双胍(山东科源制药有限公司,批号:P031-1309012)。

Fig.1 Analysis of sources of impurities related to Alogliptin benzoate in alogliptin benzoate and metforminhydrochloride tablet 图 1 苯甲酸阿格列汀二甲双胍片中苯甲酸阿格列汀部分杂质来源分析

Table 1 Summary of impurity information表1 杂质信息汇总
对照品:苯甲酸阿格列汀对照品(江苏豪森药业集团有限公司自制,批号:RS20140102,纯度 99.8%),盐酸二甲双胍对照品(中国食品药品检定研究院,批号:RS20131118,纯度 99.9%),苯甲酸阿格列汀有关物质系统适用性对照品(江苏豪森药业集团有限公司自制,批号:20150505,主要含苯甲酸阿格列汀。按面积归一化法计算(扣除苯甲酸峰),含阿格列汀不得少于 90.0 %,含杂质 D、E、F、I 与 J 均不得低于 0.2 %,各杂质在研究过程均采用 1H-NMR 和 ESI/MS 进行了结构确证)。
试剂:三氟乙酸(色谱纯,TEDIA 公司),乙腈(色谱纯,MERCK 公司),纯化水(江苏豪森药业集团有限公司自制),盐酸(AR,国药集团化学试剂有限公司),氢氧化钠(AR,国药集团化学试剂有限公司),过氧化氢(AR,国药集团化学试剂有限公司)。
辅料:甘露醇(广西南宁化学制药有限责任公司),微晶纤维素(Asahi Kasei Corporation),聚维酮 K30(BASF Corporation),交聚维酮(BASF SE),硬脂酸镁(辽宁奥达制药有限公司),薄膜包衣预混剂(胃溶型,上海卡乐康包衣技术有限公司)。
制剂样品:苯甲酸阿格列汀二甲双胍片(江苏豪森药业集团有限公司,批号:ZS0402、ZS0604、ZS0605、ZS0606),KAZANO(日本武田药品株式会社,批号:C19819,规格:12.5mg∶500 mg;批号:C19973,规格:12.5 mg∶1 000 mg)。
1.2 仪器
高效液相色谱仪(Agilent Technologies 公司),XS105DU 电子分析天平(梅特勒-托利多仪器有限公司),电热恒温鼓风干燥箱(上海精宏实验设备有限公司),药品强光照射试验箱(重庆永生实验仪器厂)。
2 方法与结果
2.1 溶液配制
取本品细粉适量(约相当于阿格列汀 25 mg),精密称定,置 50 mL 量瓶中,以水-乙腈(90∶10)为稀释液,加稀释液约 10 mL,超声 5~10 分钟,使阿格列汀溶解。再用稀释液稀释至刻度,摇匀,过滤,取续滤液作为供试品溶液。精密量取 1 mL,置 200 mL 量瓶中,用稀释液稀释至刻度,摇匀,作为对照溶液。
2.2 空白辅料配制
复方片处方含有甘露醇、微晶纤维素、聚维酮 K30、交联聚维酮、硬脂酸镁及薄膜包衣预混剂。空白辅料处方参考复方片处方进行配制,配制工艺采用粉末直接过筛混合,混合过程按照“等量递加稀释法”进行。
2.3 有关物质方法确定
2.3.1 色谱条件的选择
(1)流动相与检测波长选择
参考参比制剂专利[9]中的色谱条件,流动相 A:纯化水∶乙腈∶三氟乙酸=1900∶100∶1(V/V);流动相B:纯化水∶乙腈∶三氟乙酸=100∶1900∶1(V/V),采用反相色谱法,以氰基硅烷键合硅胶为填充剂,二极管阵列检测器(DAD:190~400 nm)上对样品进行扫描,选取280 nm 作为苯甲酸阿格列汀的检测波长(见图 2)。

Fig.2 Sample scanning results of diode array detector (DAD: 190~400 nm)图2 二极管阵列检测器样品扫描结果(DAD:190~400 nm)
以280 nm 为检测波长,流速每分钟 1.0 mL,柱温 25 ℃,以 0.05 % 三氟乙酸-水为流动相A(V/V),以 0.05 % 三氟乙酸-乙腈为流动相 B(V/V),以不同的梯度程序对苯甲酸阿格列汀进行检测分析,进行方法学研究。
(2)梯度选择
在上述流动相体系下考察不同梯度对苯甲酸阿格列汀及可能存在的杂质(杂质 A、C、D、E、F、G、I 和 J)的分离情况的影响。结果显示,在 Ⅲ 梯度条件下,主成分保留时间合适、峰形较好、各杂质间分离度符合要求。故选择此梯度进行下步实验(见表 2)。
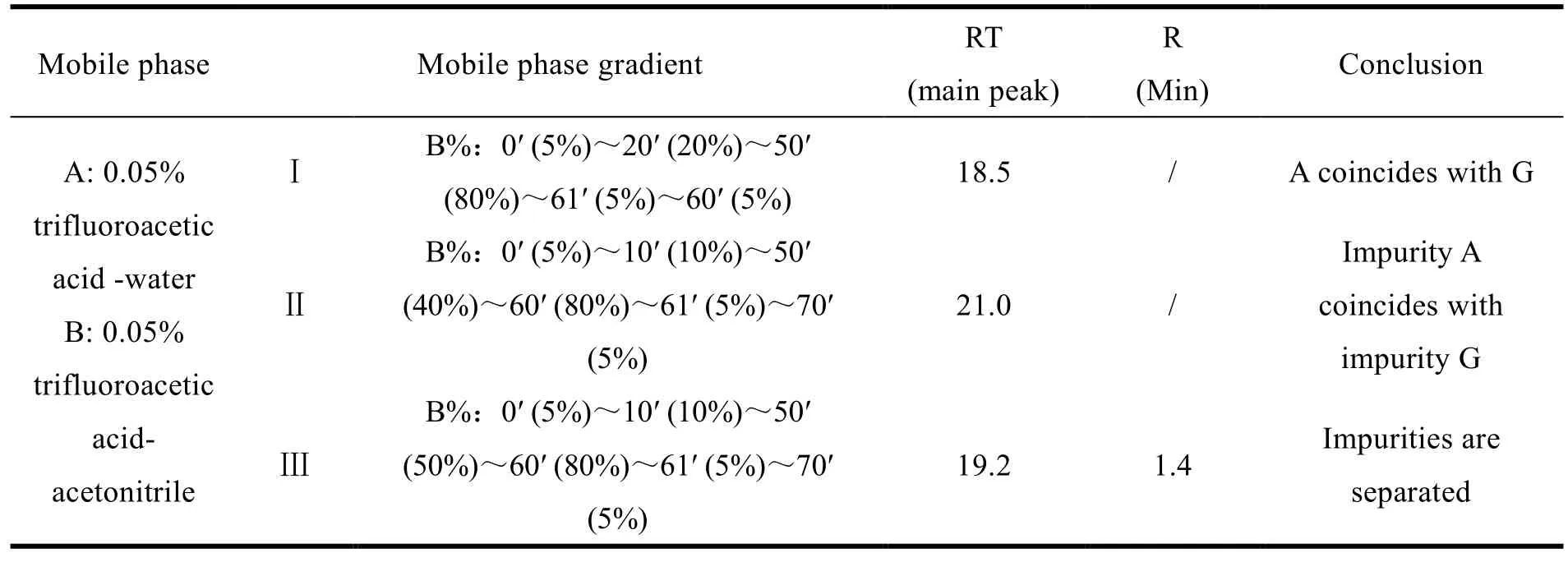
Table 2 Selection of mobile phase gradient表2 流动相梯度的选择
(3)柱温选择
选用上述确定的色谱条件,考察不同柱温对阿格列汀、苯甲酸、杂质 A、C、D、E、F、G、I 和 J 的分离效果的影响。结果显示,在 25~40 ℃ 之间,随着柱温的升高,主峰保留时间逐渐提前,相邻杂质间的最小分离情况变好。综合考虑,选用 35 ℃ 作为检测温度(见表 3)。
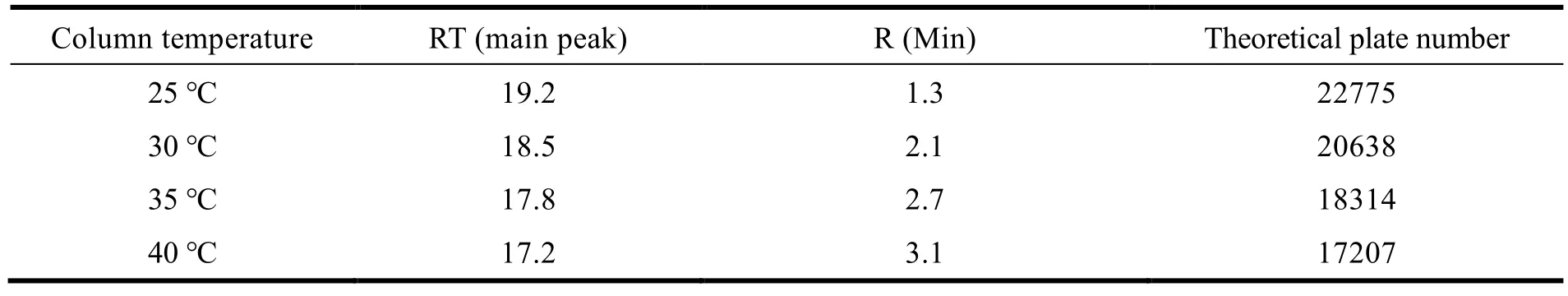
Table 3 Selection of Column temperature表3 柱温选择
根据以上实验结果确定色谱条件为:照高效液相色谱法(《中国药典》2020 年版四部通则0512)测定,以氰基硅烷键合硅胶为填充剂(Agilent SB CN,250 × 4.6 mm,5 μm 或效能相当的色谱柱),以 0.05 % 三氟乙酸-水(取三氟乙酸 0.5 mL,加水 1 000 mL 溶解)为流动相 A,以0.05 % 三氟乙酸-乙腈(取三氟乙酸 0.5 mL,加乙腈 1 000 mL 溶解)为流动相 B,流速为每分钟 1 mL,柱温为 35 ℃,按表 4 进行线性梯度洗脱,检测波长为 280 nm。

Table 4 Elution procedure表4 洗脱程序
2.3.2 方法验证
(1)系统专属性试验
为确定在上述色谱条件下,合成中的起始原料 A(杂质 A)、主要中间体(杂质 C)、已知杂质(杂质 D、E、F、G、I 和 J)以及空白辅料(包括盐酸二甲双胍)能够被明确的区分,进行方法的专属性考察。
结果显示,在该色谱条件下,盐酸二甲双胍和空白辅料对本品杂质的检测没有影响(见图 3)。苯甲酸阿格列汀与其他可能存在的起始原料、中间体及各已知杂质等的分离度均符合规定(见表5 及图 4)。所以,在确定的色谱条件下能准确的检测样品的杂质,方法专属性较好。

Table 5 The specificity and system suitability表5 专属性与系统适用性

Fig 3 Chromatogram of blank excipients (containing metformin hydrochloride)图3 空白辅料(含盐酸二甲双胍)的色谱图
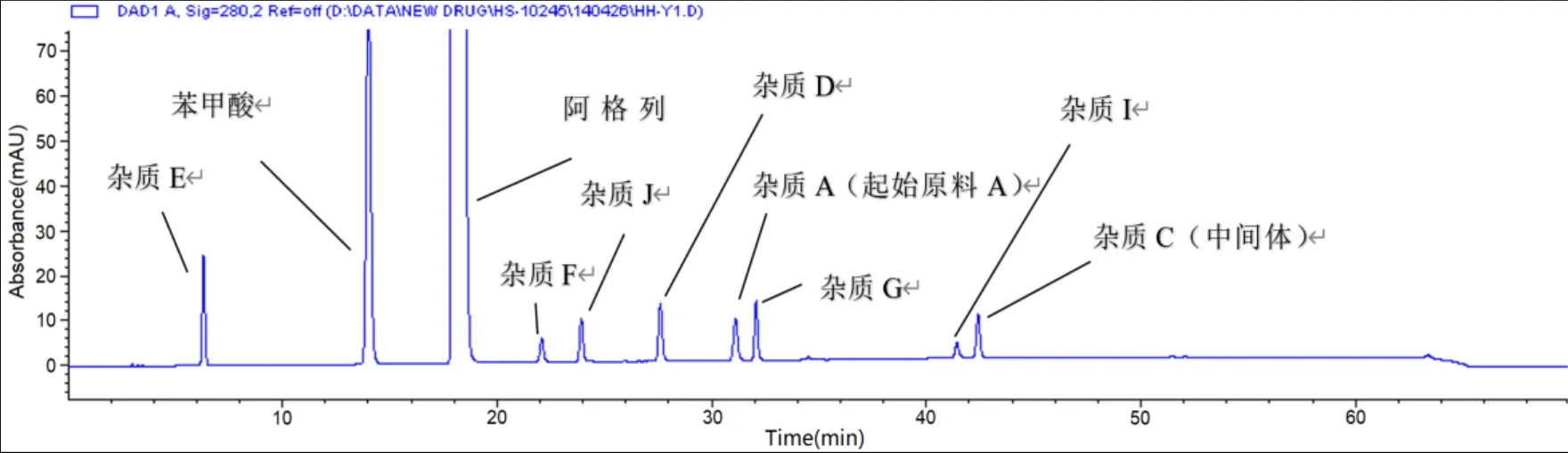
Fig.4 Separation degree of alogliptin from benzoic acid and impurities图4 阿格列汀与苯甲酸及杂质的分离度
(2)破坏性试验
取盐酸二甲双胍+空白全辅料,分别进行强酸(0.1 mol·L-1)、强碱(0.1 mol·L-1)、氧化(10% H2O2)、热解(90 ℃)、高温(105 ℃)和光照(光强度 4 500 lx ± 500 lx)破坏试验。24小时后,用稀释液配制成含盐酸二甲双胍 20 mg·mL-1的溶液。复方片适量,同上述破坏试验条件,24 小时后,配制成含阿格列汀 0.5 mg·mL-1的溶液。上述溶液分别进样,考察在该色谱条件下复方片的有关物质与降解杂质的分离和检测能力,以及盐酸二甲双胍+空白全辅料对复方片中苯甲酸阿格列汀有关物质检测的影响。由表 6 可以看出,在本色谱条件下,各强降解条件下峰纯度结果均符合规定,破坏试验所产生的降解产物能够被很好地分离、检测,空白全辅料+盐酸二甲双胍及其破坏试验所产生的降解产物不干扰苯甲酸阿格列汀有关物质的检测。复方片在热解条件下严重降解,主要降解杂质为 RRT 1.42、RRT 1.44(分子量均为 382);氧化条件下有降解,主要降解杂质为 RRT 0.59(分子量约为 357);碱条件下有降解,主要降解杂质为 RRT 0.36(杂质 E)、RRT 0.59(分子量约为 357);高温条件有降解,降解产物主要 RRT=1.44,1.53,1.59,2.27,杂质 C(见表6)。
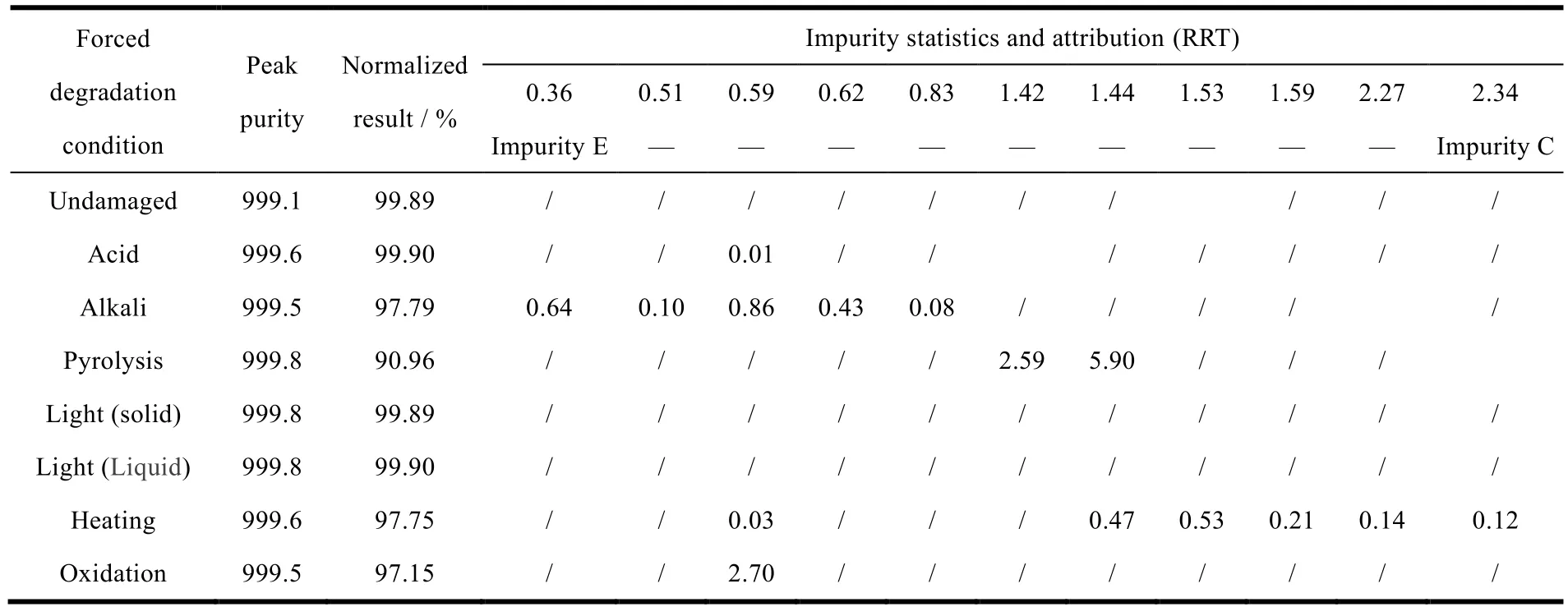
Table 6 Summary of degradation products of alogliptin benzoate and metformin hydrochloride tablets表6 苯甲酸阿格列汀二甲双胍片强制降解试验
本品在稳定性放样过程中未发现有明显的未知降解杂质产生。因此,我们暂未对破坏实验产生的杂质进一步研究。
综合上述破坏试验结果可知,采用上述色谱条件可以测定本品中的苯甲酸阿格列汀有关物质及其降解产物。
(3)检测波长的确定
在二极管阵列检测器(DAD)上提取到苯甲酸阿格列汀及主要杂质的光谱图(见图 5)。根据各已知杂质的光谱图,分别于 220 nm 和 280 nm 处比较成品及强降解实验中杂质的检测能力。
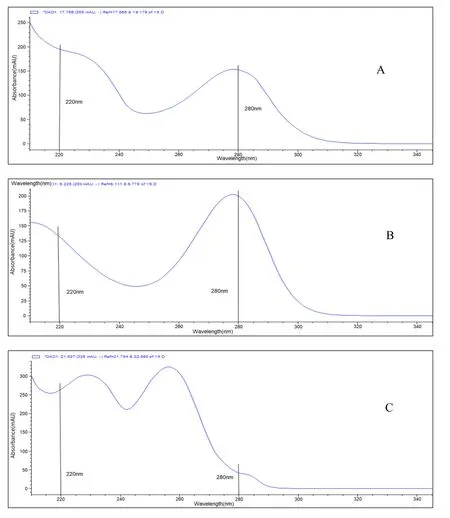
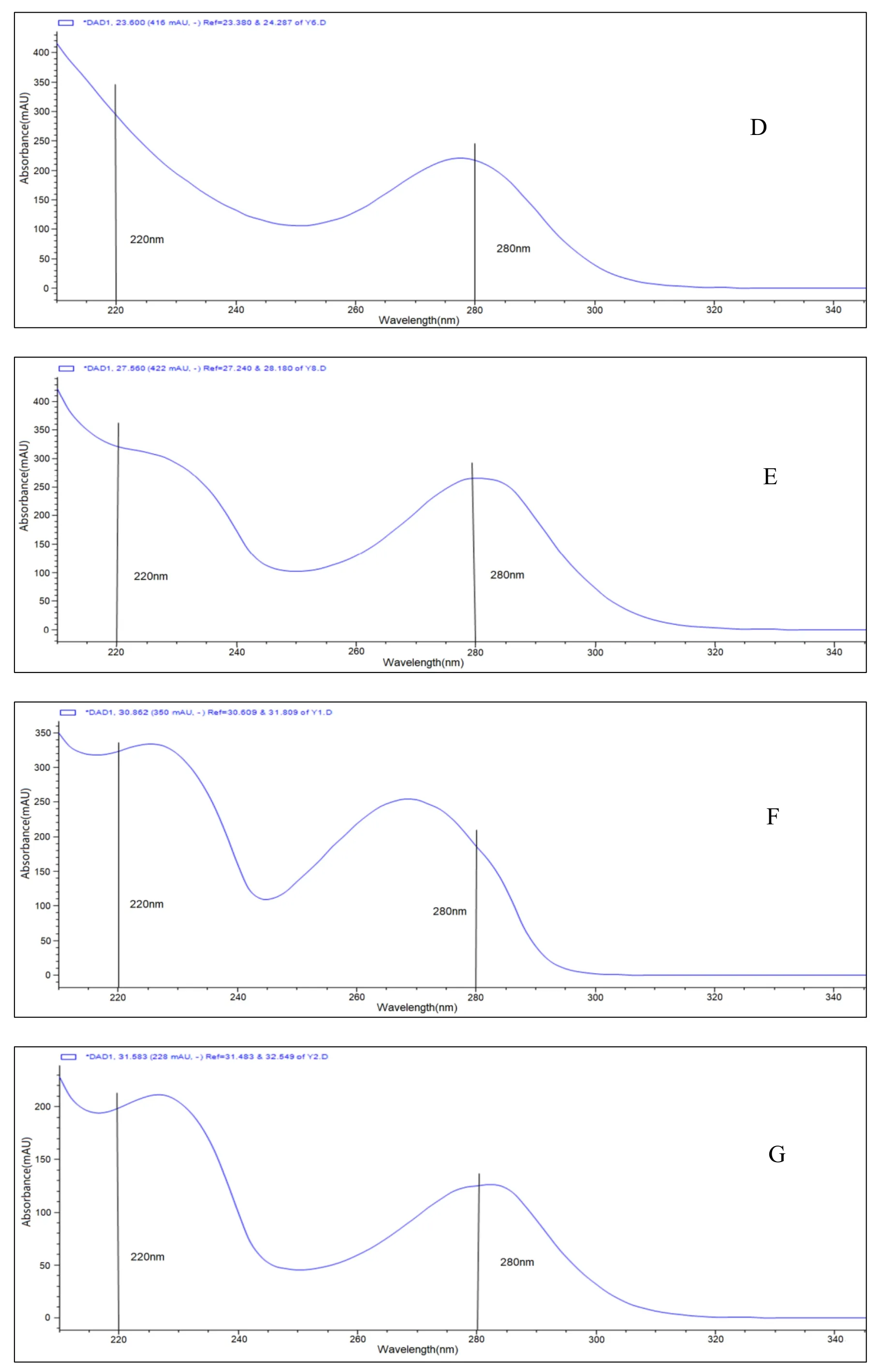

A: The spectrum of alogliptin benzoate;B: The spectrum of Impurity E;C: The spectrum of Impurity F;D: The spectrum of Impurity J;E: The spectrum of ImpurityD;F: The spectrum of Impurity A;G: The spectrum of Impurity G;H: The spectrum of Impurity I;I: The spectrum of Impurity CFig 5 The spectrum of alogliptin benzoate and the main impurities extracted on the diode array detector (DAD)A:苯甲酸阿格列汀光谱图;B:杂质 E 的光谱图;C:杂质 F 的光谱图;D:杂质 J 的光谱图;E:杂质 D 的光谱图;F:杂质A 的光谱图;G:杂质 G 的光谱图;H:杂质 I 的光谱图;I:杂质 C 的光谱图图5 在二极管阵列检测器(DAD)上提取到的苯甲酸阿格列汀及主要杂质的光谱图
从对成品及强降解实验样品的检测结果可以看出,各条件下,280 nm 检出杂质的个数高于220 nm,两者检出的未知杂质总量相当(见表 7)。综合考虑,选 280 nm 作为检测波长。nm

Table 7 The ability of different wavelengths to detect impurities in finished products and strong degradation experiments表7 不同波长对成品、强降解实验中杂质的检出能力
(4)检测限与定量限
在本试验条件下,配制不同浓度的样品溶液,分别进样确定本品与各杂质的检测限和定量限。数据结果见表 8。

Table 8 Validation data for the limit of detection and limit of quantification表8 检测限和定量限的验证数据
(5)线性范围和相对校正因子
为了消除波长对杂质检测的差异性,进行相对校正因子的测定。分别精密称取苯甲酸阿格列汀、杂质 E、F、J、D、A、G、I 及 C 约 25 mg,加乙腈溶解稀释,配制成约 5 μg·mL-1的溶液,作为杂质储备液。分别精密量取上述杂质储备液 1、2.5、5、10 和 20 mL 分别置 50 mL 量瓶中,用稀释液稀释至刻度,作为杂质限度 0.02%、0.05%、0.1%、0.2% 和 0.4% 的供试品溶液。精密量取上述溶液各 20 μL,分别注入液相色谱仪中,记录色谱图,量取峰面积。以浓度为横坐标,峰面积为纵坐标进行线性回归,以阿格列汀检测浓度(0.5 mg·mL-1)为 100% 的相对浓度,实验结果见表9。苯甲酸阿格列汀各杂质在 0.03~2.21 μg·mL-1的范围内线性关系良好,杂质 J、D、A、G 和 C在选定色谱条件下的吸收强度与苯甲酸阿格列汀相当。
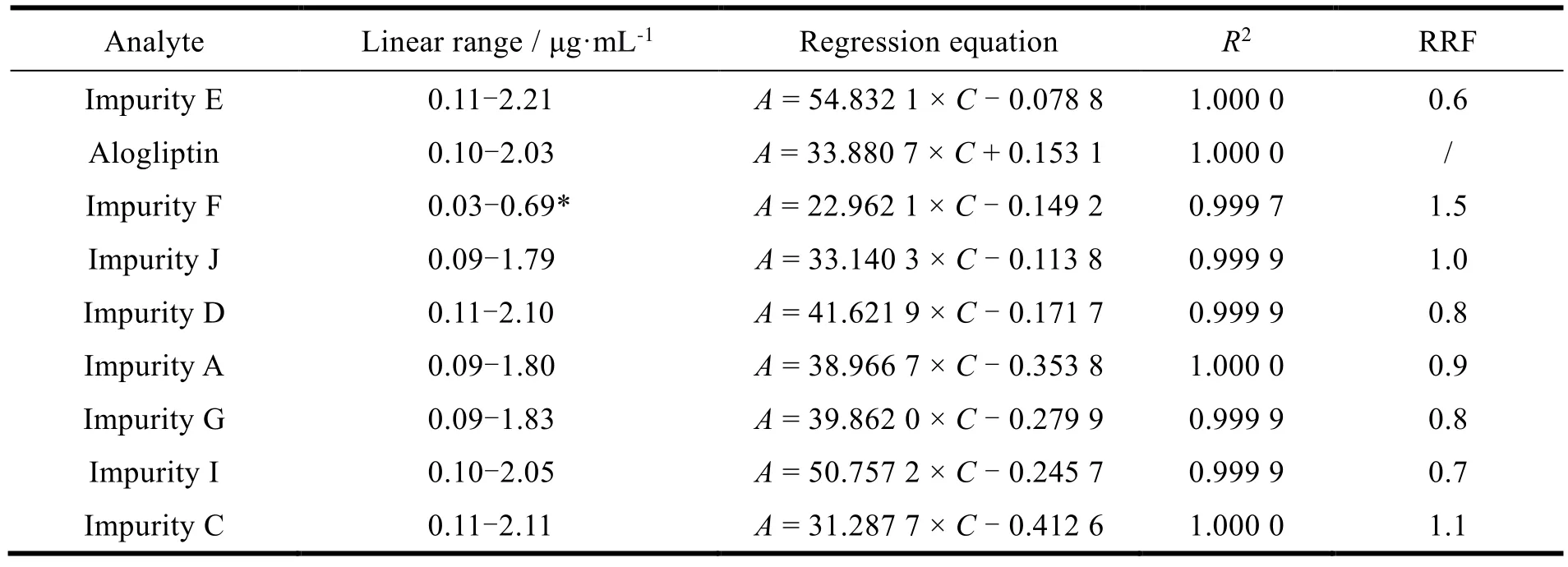
Table 9 Validation data for the linearity and relative response factor表9 线性和相对响应因子的验证数据
同时,根据各杂质与阿格列汀的斜率,采取以下公式计算相对校正因子:
杂质 F 的相对校正因子为 1.5,不在 0.8~1.2 范围内,杂质 F 在结果中乘以相对校正因子后得最终结果,可以准确反映样品杂质的质量情况。
杂质 E 和杂质 I 的相对校正因子为 0.6 和 0.63,不在 0.8~1.2 范围内,为了更好的控制本品的质量,计算均不作修正。
(6)溶液稳定性
供试品溶液在室温条件下放置 24 小时,在有关物质方法下于不同时间测定,杂质 E、F、A、G、I 和 C 均未检出;杂质 J 的平均值为 0.01 %,SD=0;杂质 D 的平均值为 0.05 %,SD=0;最大未知单杂平均值为 0.04 %,SD=0.005 %;总杂平均值为 0.22 %,SD=0.005 %。
苯甲酸阿格列汀有关物质测定溶液在室温条件下放置 24 小时稳定。
(7)杂质回收率
精密称取复方片适量,用稀释溶液溶解并定量稀释制成每 1 mL 约含阿格列汀 0.5 mg 的溶液,作为回收率基质样品。分别精密称取杂质 E、F、J、D、A、G、I 及 C,用乙腈超声溶解,配制成 0.5 μg·mL-1的溶液,作为杂质对照品溶液,平行配制 2 份。分别精密称取杂质 E、F、J、D、A、G、I 及 C 各适量至 250 mL 量瓶中,乙腈超声溶解并稀释至刻度,摇匀。精密量取 10 mL至 200 mL 量瓶中,用稀释溶液溶解并稀释至刻度,摇匀,作为母液备用。分别精密量取上述母液 1、2.5、5、10、20 mL 至含基质样品的 50 mL 量瓶中,用稀释溶液稀释至刻度,摇匀,作为回收率 0.02 %、0.05 %、0.1 %、0.2 %、0.4 % 的供试品溶液,平行配制 3 份。精密量取上述溶液各 20 μL,分别注入液相色谱仪中,记录色谱图,量取峰面积。按外标法以峰面积计算各杂质的回收率。
结果显示,在 0.02 %~0.40 % 浓度范围内,杂质平均回收率分别为 99.7 %、98.5 %、100.7 %、98.4 %、98.7 %、99.6 %、98.5 % 和 100.0 %,RSD 分别为 1.7 %、2.5 %、1.3 %、1.8 %、1.7 %、1.2 %、0.78 % 和 2.0 %(n=15),表明该方法检测各杂质准确、可靠。
(8)有关物质重复性试验
取成品,按照有关物质测定的方法重复测定 6 次,杂质 J 的平均值为 0.01 %,SD=0;杂质 D 的平均值为 0.04 %,SD=0;其他已知杂质均未检出;最大未知单杂平均值为 0.03 %,SD=0.004 %;总杂平均值为 0.16 %,SD=0.004 %。因此,说明本方法重复性很好。
(9)方法的耐用性
在本色谱条件下,微调柱温、流动相中三氟乙酸浓度以及更换不同批次的色谱柱(Agilent SB CN,250 × 4.6 mm,5.0 μm),检测系统适用性溶液和成品考察方法的耐用性,试验设计见表 10。由结果可知,各个已知杂质与主峰分离度、杂质间的分离度均较好。故本方法在色谱条件有微小变化时的耐用性良好。

Table 10 Results of robustness表10 方法耐用性
(10)中间精密度试验
取成品由人员 A、B、C 在不同时间分别用不同仪器(Agilent 1260 系列 1 和 Agilent 1200 系列)所测得的 6 个样品中,杂质 E、F、A、G、I 和 C 均未检出;杂质 J 的平均值为 0.02 %,SD=0.005 %;杂质 D 的平均值为 0.04 %,SD=0.004 %;最大未知单杂平均值为 0.03 %,SD=0;总杂平均值为 0.16 %,SD=0.01 %。
本方法通用性很好,不同仪器、不同时间及不同人员的差异很小。
2.4 样品测定
采用上述有关物质检测条件进行 4 批自制制剂与 2 批参比制剂检测,结果如表 11 所示。杂质 E、F 和 I 均未检出,杂质 J 在自制制剂中有检出,杂质 D 为降解杂质,在自制和参比制剂样品中均有检出。

Table 11 Determination results of related substances of alogliptin benzoate and metformin hydrochloride表11 苯甲酸阿格列汀二甲双胍片有关物质测定结果
2.5 限度制定考虑
结合表 10 自制制剂和参比制剂检测结果以及产品稳定性研究结果(未发表数据),对于已知杂质只检出了杂质 J 和 D。其中杂质 D 为潜在的降解杂质,同时结合 ICH Q3B[10],规定其限度为 0.5 %,规定杂质 J 的限度为 0.2 %。
其他已知杂质均未检出,其中杂质 E 为潜在的降解杂质(制剂中碱破坏条件降解),杂质 F为主峰后杂,杂质 I 为原料药中较难去除的杂质。因此,将杂质 E、F 和 I 订入质量标准,限度均为 0.2 %。
按一般制剂对杂质控制的限度要求,同时结合各批样品的检测结果,规定其它单个未知杂质的限度均为 0.2 %,总杂限度为 2.0 %。
3 讨论
本研究结合原料药的合成路径以及复方片可能存在的降解途径,充分的分析了相关杂质的来源,结合《中国药典》、ICH Q2[11]系列杂质研究的相关指导原则,对复方片中的杂质检测方法进行了充分的验证,最终结合自制制剂和参比制剂的检测结果合理提出杂质控制限度。该方法已成功应用于本品后续的稳定性检测、生产质量控制中。
同时,糖尿病治疗的临床用药便捷性的需求在不断增加[12-13],复方制剂是未来的发展方向之一。本品杂质控制方法的研究,也为后续含有苯甲酸阿格列汀的复方产品质量控制提供一种样品处理便捷、重复性好、精密度高的检测方法。
猜你喜欢
民间文学(2021年11期)2021-03-31
云南化工(2020年11期)2021-01-14
民间文学(2020年8期)2020-08-31
安徽医科大学学报(2016年12期)2017-01-15
百科知识(2016年22期)2016-12-24
国外医药(抗生素分册)(2016年1期)2016-07-10
广东第二课堂·小学(2016年6期)2016-05-14
作文评点报·作文素材小学版(2016年16期)2016-05-06
当代化工研究(2016年5期)2016-03-20
中国继续医学教育(2015年3期)2016-01-06
