
烟气单质汞催化氧化技术研究进展
2023-06-13 13:03李国良叶凯航耿孟达
能源环境保护 2023年3期
李国良, 叶凯航, 耿孟达, 郑 扬, 岳 涛
(北京科技大学 能源与环境工程学院, 北京 100083)
0 引 言
汞,俗称水银,化学符号为Hg,是一种常温下可以以气态和液态形式存在重金属。汞在烟气中主要存在三种形态:气态元素汞(Hg0)、气态氧化汞(Hg2+)和颗粒态汞(Hgp)。在大气环境中汞主要以Hg0形态存在,它可以在大气中停留数月至一年时间,可以随气流传输到远离排放源的地区,导致环境中汞浓度的增加,并对健康和经济造成不可估量的损害[1-3]。因此,汞及其化合物作为全球性污染物受到研究人员的广泛关注。
2001年起,联合国环境规划署(United Nations Environment Programme, UNEP)发表了《全球汞评估报告》(Global Mercury Assessment),对全球汞排放情况做了详细评估[4]。为了在全球范围内有效减少汞的释放、使用和排放,减少汞及其化合物损害生态环境和人类健康,国际社会于2013年编写了具有法律约束力的汞文书,生成《关于汞的水俣公约》,该公约于2017年8月16日生效。公约要求各缔约方控制并于施行时减少大气汞排放。根据联合国环境规划署(UNEP)的汞排放数据,全球主要国家的汞排放达到1 431 t,化石燃料燃烧占49.9%[5-7]。我国是全球大气汞排放量最大的国家,2017年,我国大气汞人为排放量为444 t,大约占全球大气汞排放量的30%,所以我国汞控制排放对全球汞减排具有重要意义[8]。因此,我国大气汞排放控制也得到全球的广泛关注,面临巨大的履约压力。
汞化合物在燃烧过程中完全转化为Hg0,随着温度的降低,部分Hg0转化为Hg2+和Hgp[9]。汞的化学形态对空气污染控制装置(APCDs)的脱除效率具有显著影响。湿法烟气脱硫(WFGD)可以捕获Hg2+,静电除尘器(ESP)、布袋除尘器(FF)等除尘器可基本去除Hgp污染物。由于高挥发性和低水溶性,Hg0是最难捕获的化学形态[10]。以燃煤电厂为例,目前燃煤电厂的污控措施基本都具有选择性催化还原催化剂(Selective Catalytic Reduction,SCR)或是选择性非催化还原脱硝措施,布袋除尘或是电除尘设备,WFGD以及湿式电除尘,如图1所示。
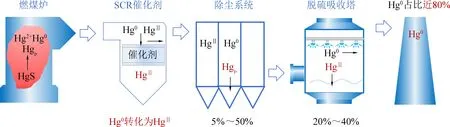
图1 燃煤电厂污控设备流程图Fig. 1 The flow diagram of air pollution control systems in a coal-fired power plant
随着我国对NOx和SO2控制加严,燃煤工业锅炉和水泥行业也逐渐增加脱硝设备和湿法脱硫设备。在此污控条件下,我国计划采用多种污控设施协同控制的方法降低烟气中汞排放[6]。烟气中,99%以上Hgp可以被除尘设备脱除,80%以上Hg2+可以被湿法脱硫设备脱除,但是Hg0很难被污控设备直接捕获下来[11-12]。在燃煤烟气中,Hg0大约占到了烟气HgT的20%左右,在褐煤等低品位煤炭烟气中,Hg0可以占总汞排放的比例高达80%以上[13]。因此,Hg0的去除是解决燃煤烟气汞排放的关键问题。通过催化剂将Hg0转化为Hg2+,再通过脱硫设备捕获Hg2+,从而依靠现有设备协同控制有效地减少Hg0排放是控制汞排放的理想技术途径。因此,目前协同技术关键步骤是制备高效Hg0氧化催化剂。
综上所述,随着我国重金属污染防治的重大需求和《关于汞的水俣公约》行动计划的逐步推进,我国未来可能通过采用催化氧化和湿法脱硫协同控制燃煤烟气汞排放。Hg0催化氧化是该协同控制技术的关键步骤。目前,Hg0氧化催化剂可以归为三个大类:钒基脱硝催化剂、贵金属氧化物催化剂和过渡金属氧化物催化剂[11, 14-21]。
1 烟气汞催化氧化技术
1.1 钒基催化剂对Hg0的氧化研究
钒基催化剂是选择性催化还原技术中用于将氮氧化物(NOx)还原为氮氧化物(N2)的催化剂。钒基催化剂氧化汞的机理尚不完全清楚[22-23]。一般认为,SCR催化剂对NOx还原和对Hg0氧化分别在两个区域进行。如图2所示,钒基催化剂一般包括富氨区和贫氨区。NOx一般在富氨区还原,由于NH3可以与Hg0竞争活性位点,Hg0在该区域的氧化受到抑制。在NH3贫氨区,NH3含量因与NO反应而减少,该段为Hg0主要氧化位置。

图2 SCR催化剂汞氧化过程示意图[14]Fig. 2 Mercury oxidation mechanism of SCR catalysts[14]
SCR催化剂主要是以钒氧化物(V2O5)作为活性物质,钒基催化剂表面的活性氧作为化学氧化位点吸附并氧化Hg0为Hg2+[24]。研究表明,烟气中的HCl对Hg0的氧化性能起到关键作用。He[25]使用V2O5-WO3/TiO2催化剂对Hg0进行催化氧化测试,HCl添加使催化剂对Hg0的氧化效率达到65%。HCl对Hg0氧化的促进机理主要包括两种机制:迪肯制氯(the Deacon process)和表面活性氯。
(1)迪肯反应指HCl在催化剂表面与O2反应形成Cl2和H2O[26],反应机制如下式所示:

(1)
(2)
(3)

(4)
在实际烟气中,SCR反应中NH3会强烈抑制Hg0发生竞争吸附与氧化,由于NH3浓度远远高于Hg0浓度,从而严重抑Hg0的催化氧化效率,这是商业钒基催化剂汞氧化性能面临的重要问题。另外,商业催化剂汞氧化严重依赖HCl形成的活性位点,我国煤炭中Cl含量相对较低。商业SCR催化剂对Hg0的催化氧化具有一定的活性,但是这种活性严重依赖HCl形成的Cl2或活性Cl,因而随着烟气组分和运行条件不同而表现出较大差异。同时,V2O5作为单一活性位点难以同时满足NO还原和Hg0氧化的要求。因此,大量研究采用Mn、Ru、Ag、Ce等元素改性V2O5-WO3/TiO2,构建双活性反应中心,促进催化剂NO还原与Hg0氧化协同反应。
如图3所示,段钰锋团队采用Mn改性VWTi催化剂,Mn元素的引入显著提高VWTi催化剂低温(120~200 ℃)Hg0氧化效率从65.0%±16.7%到98.0%±1.1%,但是在SO2和H2O条件下,Mn-VWTi的Hg0氧化被严重抑制。晏乃强[27-28]团队采用Ru、Ag、Ir等贵金属元素作为活性剂掺杂脱硝催化剂,良好的催化活性可以使Hg0完全转化为Hg2+,同时将NH3直接氧化为氮气,从而避免NH3对Hg0氧化抑制作用,但是NH3单独存在条件下依然严重抑制催化剂的Hg0氧化性能。李彩亭团队[11]采用CeO2改性SCR催化剂使其Hg0氧化效率从42.6%提升到88.9%,并且探究了各种烟气组分对V0.80WTiCe0.25催化剂的催化效果影响,SO2和NH3降低催化剂的氧化效果到41.8%~49.7%,NO促进了催化剂的Hg0氧化效率达到75.1%。因此,双活性位点的构建促进了钒基催化剂的Hg0氧化效率,同时SO2、NH3等烟气组分的抑制作用依然存在,未来需要进一步提高催化剂的抗硫抗氨性能。
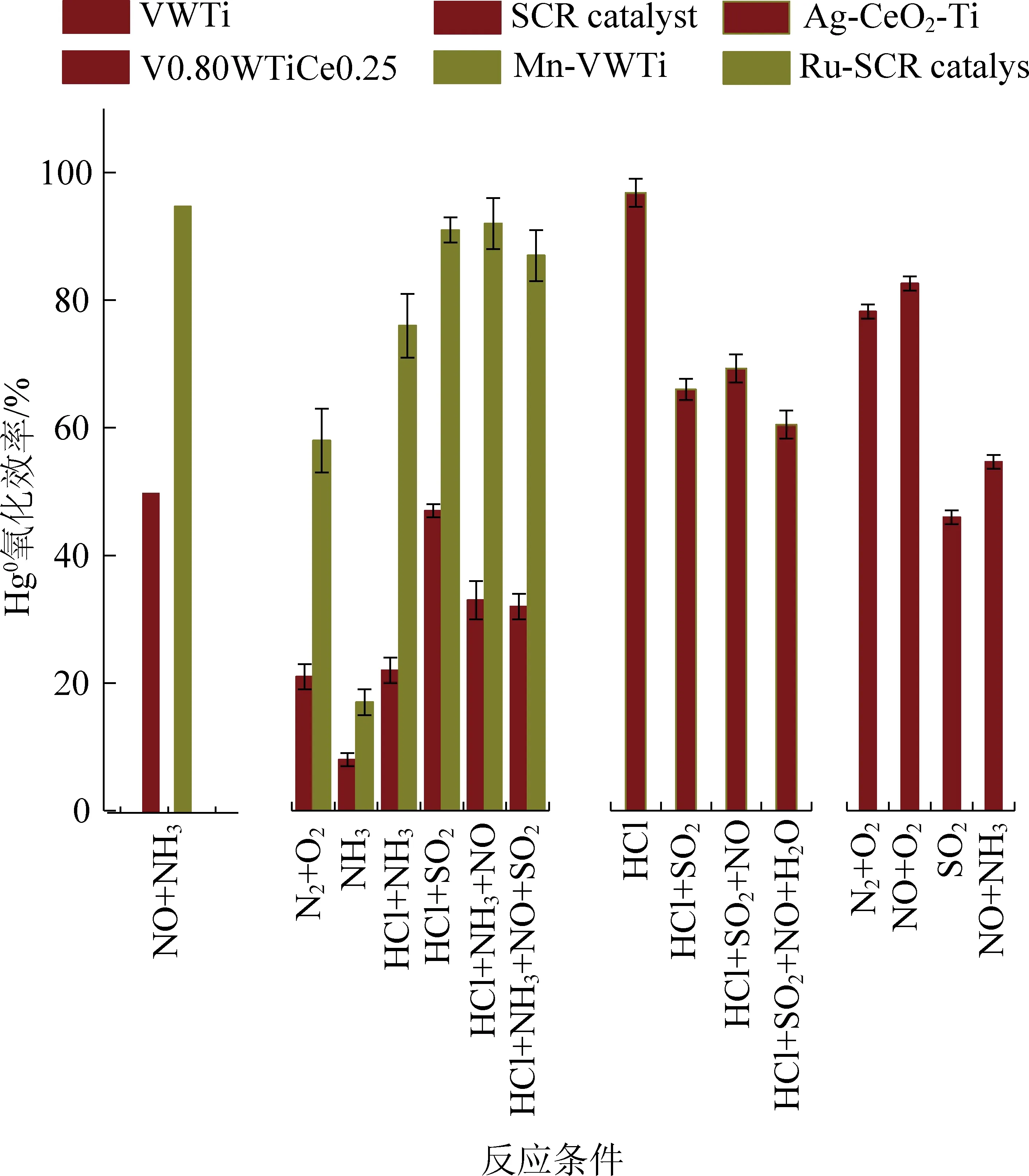
图3 改性SCR催化剂的汞氧化性能[27-34]Fig. 3 Hg0 oxidation efficiency of modified SCR catalysts[27-34]
1.2 贵金属氧化物对Hg0的氧化研究
贵金属(Au、Ag、Pd、Pt等)对可以选择性吸附Hg0,形成固态合金,因此在贵金属催化剂对Hg0氧化应用方面具有良好的前景[16, 28, 35-39]。Presto[40]在中试条件下探究了Au、Pd和Pt贵金属的催化剂以及烟气组分影响,结果表明:Pd和Pt基催化剂随着时间推移对Hg0氧化效率明显下降,而Au基催化剂保持了良好Hg0氧化性能。Pd和Pt基催化剂失活归咎于长时间的O2环境暴露使Pd和Pt原子转化为氧化物,在HCl缺失的条件下,Hg0无法进一步转化为Hg2+。Jason[40-43]采用一次浸渍、二次浸渍和喷涂方法制备了各种Au/TiO2,喷涂法制备催化剂因负载量高和脉冲过程中损失量少表现出最佳催化效果,Hg0氧化效率达到40%~60%。在燃煤电厂烟气治理示范过程中,半年运行时间使Au基催化剂氧化效率从80%降到77%,一年运行后降到57%。研究表明:Au基催化剂的活性下降并不是因为SO2中毒,而是由于飞灰堆积对活性Au原子的覆盖。Hamoon[44]发现Au原子对Hg0吸附性能主要依赖于Au原子簇的电荷、空穴以及其他原子掺杂相关,相较于Au阳离子而言,Hg0原子更容易吸附于Au原子位置,证明Au与Hg0原子良好的相容和成键性能。
柴立元团队探究了在高浓度(10 000 ppm)SO2有色烟气条件下Pd/CuCl2/γ-Al2O3催化剂对Hg0的氧化性能,结果表明在HCl存在条件下,催化剂对Hg0氧化效率高达87%,表现出良好的抗硫性[54]。Stephen用铂、钯硝酸溶液作为改性剂浸渍Al2O3,形成烟气Hg0吸附剂Pt/Al2O3和Pd/Al2O3。Pt和Pd在204~371 ℃范围内对气体Hg0有良好的吸附,汞吸附量随金属负载量的增加而成比例增加。Hg0吸附在Pd/Al2O3上生成了固体Pd-Hg汞齐化合物。Albert[40-41]研究了在不同HCl和O2浓度下,质量分数1%的Au、Pd和Pt负载在2 mm氧化铝珠上的汞氧化能力。实验结果表明,Pd、Pt基催化剂的反应速率和催化活性随时间的推移逐渐降低,而Au基催化剂的活性在整个实验时间内保持较高。这种失活行为归因于Pt和Pd氧化物的O2消耗导致缺乏Hg0氧化的氧化剂。
贵金属(Ru和Ir)对催化剂的氧化活性和抗SO2/NH3性能均有促进作用。晏乃强团队[28]发现钌(Ru)改性SCR催化剂在低HCl浓度下对Hg0氧化具有较高的催化活性,Ru改性明显提高了Hg0化和NH3氧化能力。此外,Hg0的氧化主要依赖于活性氯(Cl*),而不是Cl2,SO2抑制氯原子结合形成Cl2,但对Cl*的生成影响很小。晏乃强团队[28]采用溶胶-凝胶法和浸渍法改性IrO2。溶胶-凝胶法促使IrO2在催化剂表面分散性更好,因此表现出改性效果更高。一方面,IrO2改性增加了化学吸附氧的补充;另一方面,IrO2促进Cl原子结合形成Cl2和HCl生成活性Cl*,Cl2和Cl*分别与气相中的Hg0反应提高催化剂表面吸附态汞(Hgad)。这两种机制的主要区别在于吸附的Hg0和HCl是否首先被活性物质氧化。同时,IrO2的掺杂对两种氧化过程都有促进作用。
综上所述,贵金属催化剂由于特有Hg0亲和性,在低温条件下形成汞齐化合,可以作为Hg0氧化的第一步,达到一定的抗SO2和抗NH3效果;高温条件下,贵金属催化剂依然依靠活性氧或活化氯达到Hg0氧化的效果。尽管贵金属氧化物对Hg0具有良好的催化活性,但是其高昂的价格使催化剂造价过高,成为其在实际中广泛应用的重要障碍。
1.3 过渡金属氧化物对Hg0的氧化研究
过渡金属(如Cu、Mn、Fe)氧化物因为具有价格低廉、活性高的特点而被广泛用来催化氧化烟气中的Hg0。图4系统分析了Fe、Mn、Ce和Co基催化剂的汞氧化性能。Xu[55]利用HZSM-5分子筛和Fe作为载体和活性剂制备出了用于汞氧化的Fe/HZSM-5催化剂,汞氧化实验结果表明275 ℃左右具有最高的氧化效率,几乎达到100%,说明催化剂具有很高的催化氧化Hg0活性。但是,这种材料容易与烟气中SO2发生反应,形成中毒。Yang探究了CuMn/Ti催化剂对Hg0的氧化效果,结果表明在SCR环境条件下催化剂汞氧化效率达到了95%以上,但是SO2的出现使催化剂氧化效率降低到40%以下[56],这种材料也面临同样的问题:催化剂与SO2发生反应产生硫酸盐化合物占据活性氧,抑制Hg0吸附与氧化,产生SO2中毒现象。Wen等发现NO明显提高CeO2/γ-Al2O3催化剂的汞氧化性[57]。NO和O2提高Co基催化剂的汞氧化性,200~1 000 ppmNO使汞氧化效率从95%上升到98%[58]。但是相反的结果也有报道,Li等[59]发现通过添加HCl促进Deacon反应,提高了催化剂汞氧化效率,但是NO在无氧条件下由于竞争作用,抑制了Hg0的氧化。这是因为无氧条件下NO无法转化为汞氧化位点NO2[55, 60-61]。综上所述,Cu、Mn和Fe等过渡金属氧化物在低温窗口对Hg0有很强的催化氧化活性,但是这种活泼的金属氧化物同样容易与SO2发生反应而中毒,并且在中高温温度窗口氧化活性较差。
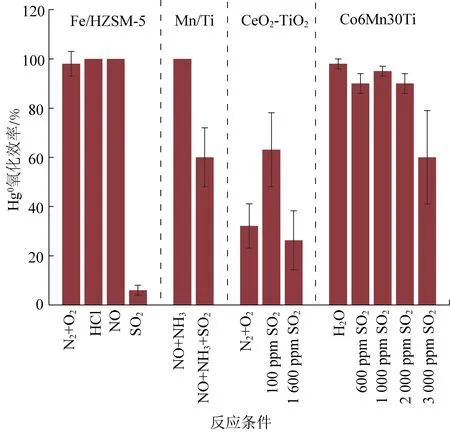
图4 过渡金属氧化物催化剂汞氧化性能[52, 55, 58, 62-63]Fig. 4 Hg0 oxidation efficiency of transition metal oxides catalysts[52, 55, 58, 62-63]
铈氧化物具有良好的催化氧化活性和多种价态变化受到了催化领域的广泛关注[64-66]。研究发现,由于铈氧化物中Ce4+/Ce3+氧化还原电对可以通过吸氧放氧自由变换,所以铈氧化物具有较高的氧化还原活性和活性氧储存性能[66]。在铈基催化剂表面通过吸氧放氧过程形成了大量不饱和氧空穴、不饱和电子空穴、活性氧等活性位点,这些活性位点较高的氧化还原性能促进了催化氧化反应的发生[67]。研究表明铈氧化物Ce4+/Ce3+氧化还原电对产生活性空穴和活性氧的过程如下[67]:
(5)
式(5)中“□”代表活性氧空穴和电子空穴。铈氧化物表面的活性氧可以作为Hg0氧化反应活性位点,电子空穴可以吸收传递电子,促进Hg0向Hg2+的转移,氧空穴可以促进氧原子传递和活性氧的补充,这些特点均有利于Hg0氧化反应的进行[24]。
铈氧化物表现出良好的烟气脱硝活性,可以作为V元素的理想替代活性剂[68-69],但是高低温度下SO2和H2O对催化剂影响差异比较大,350 ℃以上时,SO2和H2O对催化剂的活性影响不大,这可能是高温条件下SO2和H2O吸附性能较低;低于300 ℃时,SO2和H2O对催化剂的活性抑制作用显著,并且停止添加后,催化剂活性仍然不能恢复,说明Ce基催化剂的抗硫抗水性还有待于进一步提高[68]。CeO2催化剂也被广泛用于Hg0的催化氧化并在特定温度窗口表现出良好的活性[57, 70-72]。Wen等研究了CeO2/γ-Al2O3催化剂对Hg0的催化氧化,结果显示催化剂表明在150~430 ℃区间Hg0氧化效率约为65%~85%[57]。
近些年来,研究发现Ce氧化物对Hg0具有良好的催化氧化能力[73-74]。Wang等研究了Ce-Mn/Ti催化剂对Hg0的氧化性能,结果表明催化剂在250 ℃对Hg0氧化效率超过90%,同时具有良好的脱硝表现,但是较窄的反应温度窗口和较差的抗硫性限制了其应用[73]。He等利用溶胶-凝胶法制得CeO2/TiO2-PILCs催化剂,实验发现Ce含量15%时催化剂汞氧化效率最高,HCl通过Deacon反应将Hg0氧化为氯化汞[75]。
W氧化物(WO3)被认为是SCR催化剂最为有效的促进剂。钨氧化物加入有利于提高SCR催化剂表面酸性,提高对NH3吸附能力,有利于拓宽催化剂反应温度窗口;同时WO3可以降低氨和SO2的氧化以及提高抗K性;WO3的重要作用是提高B-酸位点的酸性、强度和密度,从而提高氨的吸附和反应[76]。另外,WO3可以促进Ce的还原,稳定Ce氧化性,从而有利于提高Ce-SCR催化剂的脱硝活性。鉴于Ce和W氧化物的良好活性,已有研究将两者优点结合,利用Ce-W共氧化物促进Hg0的氧化,结果表明在HCl存在条件下CeO2-WO3/Ti催化剂在200~400 ℃温度窗口范围内对Hg0的氧化效率高于90%以上,但是抗水性较差,8%H2O添加使催化剂氧化效率下降到60%~80%,并且CeO2-WO3/Ti催化剂汞氧化活性过分地依赖HCl[70-71]。因此,尽管铈钨催化剂具有良好的活性,但是铈钨钛催化剂较差抗水性以及对HCl的依赖性,造成铈钨催化剂在HCl缺失条件下的氧化活性和抗水性还有待于进一步改善。
因此,过渡金属氧化物催化剂以其种类多样、化学性质活泼等优点可以满足于低温条件下对Hg0催化氧化的要求。但是由于过渡金属催化剂活泼的化学性质导致催化剂对SO2和NH3具有较好的吸附性,造成催化剂抗SO2和抗NH3性能较差。
综上所述,本研究在总结V基催化剂、贵金属催化剂和过渡金属催化剂的基础之上,总结Hg0催化氧化研究存在的难点包括三点:(1)NH3与Hg0竞争活性位点,单一位点无法满足脱硝和Hg0氧化协同催化;(2)SO2、H2O会与Lewis酸位点反应,导致催化剂活性位点失活,造成Hg0氧化活性降低;(3)构建Hg0催化活性位点,提高金属氧化物吸氧放氧循环能力,增加Lewis酸位点数量,提高催化剂协同脱除Hg0能力。如图5所示,本研究认为未来研究应关注在多活性位点的构建,分离不同污染物的吸附反应区域,降低污染之间竞争和干扰。Cu、Fe等金属氧化物可以作为强碱性活性位点,W、Mo等金属氧化物可以作为酸性活性位点,贵金属元素和Ce、V等元素可以作为Hg0吸附和氧化中心。通过结构优化和酸碱调控化构建酸性中心、碱性中心以及氧化中心,使碱性气体(NH3)、酸性气体(NO、SO2、HCl等)以及Hg0等污染物分别吸附在酸性中心、碱性中心以及氧化还原中心,减少竞争吸附造成的催化剂失活,达到协同催化脱除NO和Hg0多种污染物的效果。
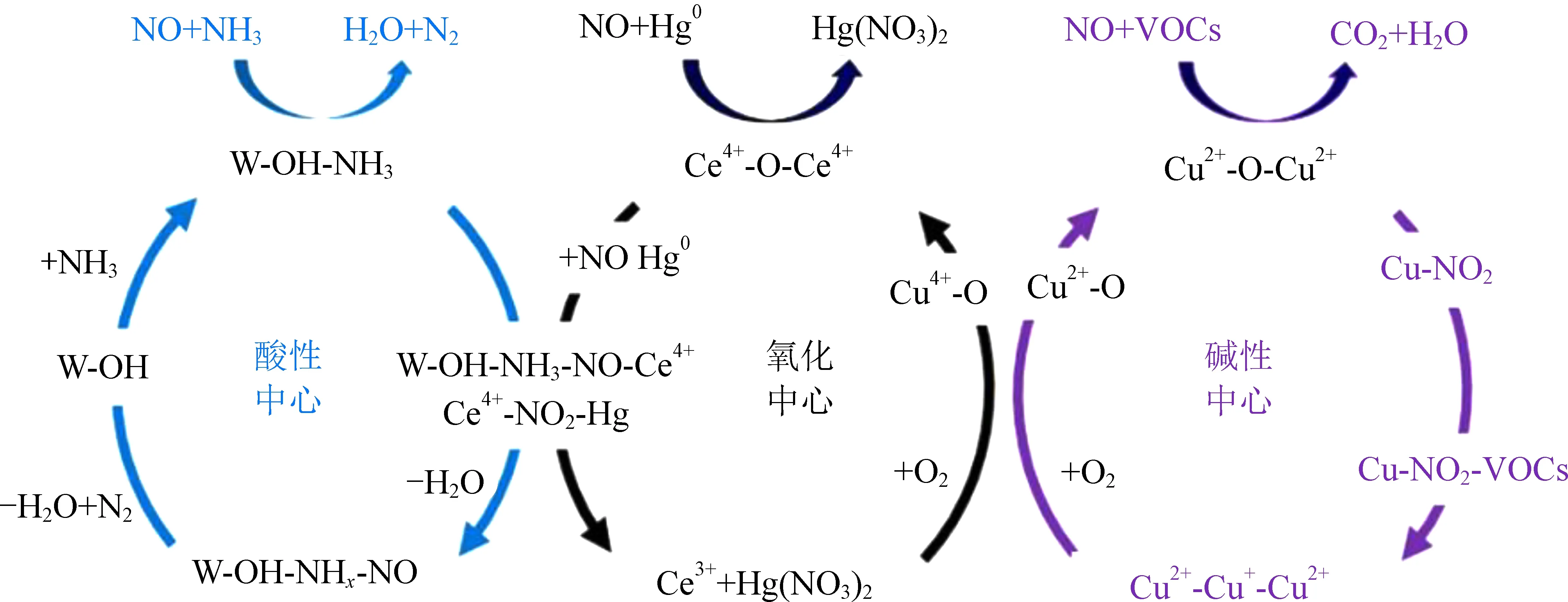
图5 多污染协同净化催化剂设计策略Fig. 5 Design strategy of multi-pollutants synergy purification catalyst
2 烟气组分相互影响机理
2.1 常规污染物影响
不同烟气组分(HCl、NO、SO2、H2O等)对汞催化氧化的影响也是学者研究的重点[55, 77]。烟气组分、浓度、运行条件等因素都可能导致催化氧化汞氧化效果差异,因此不能忽视烟气条件对催化剂氧化Hg0的影响[24]。张军营团队[59, 78]详细地研究了CeO2/TiO2型催化剂在各种烟气组分对Hg0的催化氧化性能。结果表明:Ce1.5Ti催化剂在250 ℃时Hg0氧化效率达到95%;烟气中HCl对Hg0氧化效率提高起到重要作用,主要通过迪肯制氯反应将Hg0转化为Hg2+;由于竞争吸附,烟气NO会轻微地抑制Hg0催化氧化。但是相反的结论也有报道,Wen等发现NO的出现明显提高了CeO2/γ-Al2O3催化剂汞氧化效率[57]。常化振团队[79]采用不同pH沉淀溶液制备VMo/Ti和CeMoOx催化剂,形成强碱性、中强碱和弱碱性活性位研究催化剂协同脱硝脱汞反应和中毒机制。结果表明:HCl优先与强碱性活性位反应,造成NO、NH3的SCR反应失活,但是促进了Hg0氧化为HgCl2;NH3与Hg0竞争活性位,造成Hg0氧化效率下降,研究结果建议平衡催化剂表面酸碱性有利于促进Hg0氧化效率。H2O对Hg0氧化抑制作用主要发生在低温条件下,水分子在催化剂表面形成水膜,阻碍了Hg0向催化剂表面活性位点的迁移[29];中高温条件下,H2O对Hg0氧化抑制作用相对较小[80]。
SO2是影响Ce基催化剂的重要因素[81]。当SO2浓度低于400 ppm时,CeO2/TiO2催化表面可以将SO2氧化为SO3,进一步将Hg0氧化成HgSO4,从而促进Hg0氧化效率;当SO2浓度上升到1 000 ppm时,Hg0氧化效率急剧下降,这是因为高浓度的硫氧化物与Hg0发生强烈的竞争吸附,SO2在催化剂表面占据CeO2,形成大量硫酸盐,导致活性位点永久失活,致使Hg0不能接触到活性位点,从而使氧化反应难以进行[59, 82-83]。
Senior[84]采用一个动力学模型,用于测试燃煤电厂通过SCR催化剂Hg0氧化效率,该模型考虑了多孔SCR催化剂内部的扩散以及NH3和Hg0对催化剂活性位的竞争,对8种不同的钒基催化剂均有较好的拟合效果。拟合结果表明,HCl对汞氧化具有促进作用,而NH3对汞氧化有相反作用。Kamata[85]也指出,当HCl浓度增加到4.5 ppm时,V2O5(WO3)/TiO2催化剂的汞氧化效率从80%提高到100%,而当NH3/NO比例增加到1.0时,汞氧化效率降低到零。沈伯雄团队[60-61]系统探究了NO和NH3对6Ce6MnTiP催化剂的汞氧化活性的影响。结果表明,由于NH3与Hg0的竞争吸附,NO和NH3的共存抑制了催化剂对汞的吸附。
本研究总结了NO、HCl、SO2、NH3、H2O等烟气组分对V基催化剂、贵金属催化剂和过渡金属催化剂的Hg0氧化性能影响机制[27-34, 52, 55, 58, 60-63, 79]。NO和HCl主要表现为促进催化剂Hg0氧化效率,NO和HCl在催化剂吸附位点形成-NO2、Cl*、Cl2等官能团,促进Hg0氧化为Hg(NO3)2和HgCl2[55, 60-61]。如图6所示,我们总结了关于SO2和NH3影响的研究文献[27-34, 55, 60-61],结果表明:不含SO2的烟气Hg0氧化效率为94.7%±3.9%,但添加SO2之后,Hg0氧化效率下降到66.8%±16.8%;不含NH3的烟气Hg0氧化效率为83.9%±4.8%,但添加NH3之后,Hg0氧化效率下降到57.1%±7.5%。SO2和催化剂活性位点形成硫酸盐,造成活性位点失活,NH3与Hg0对活性位点的竞争造成Hg0氧化效率,因此,SO2和NH3为催化剂Hg0性能下降的主要原因。由于SO2和NH3烟气浓度远高于Hg0,所以Hg0很难在相同活性位点竞争过程中表现出高于SO2和NH3吸附性能。基于以上结论,有效地分离SO2、NH3和Hg0的吸附反应区域,降低SO2和NH3对Hg0的竞争作用,提高催化剂的催化氧化活性,是提高催化剂对Hg0氧化效率的重要途径。
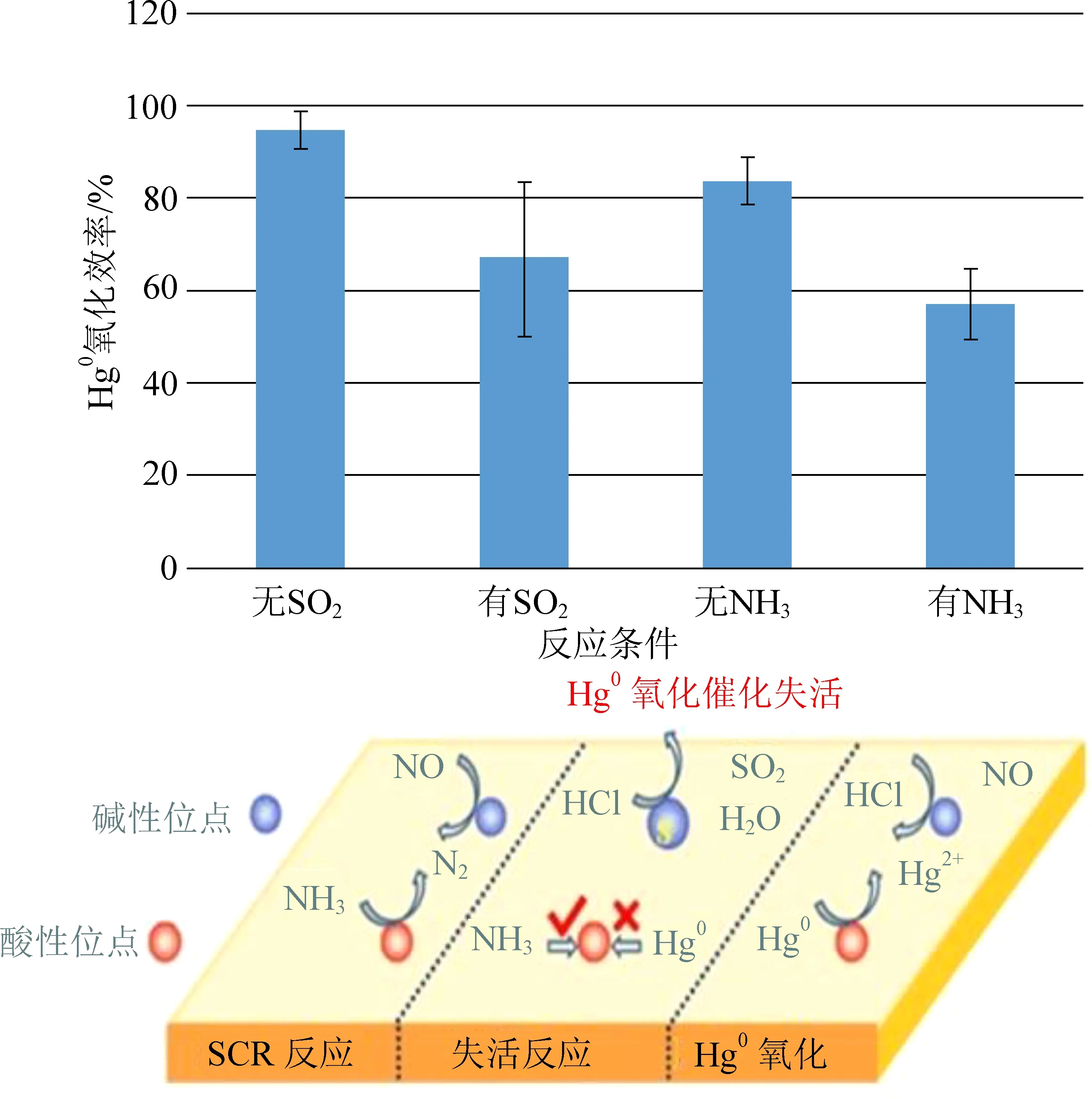
图6 Hg0氧化催化剂中毒失活机理[27-34, 52, 55, 58, 60-63, 79]Fig. 6 Poisoning and deactivation mechanism of Hg0 oxidation catalyst[27-34, 52, 55, 58, 60-63, 79]
2.2 非常规污染物影响
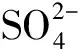
二噁英(PCCD/Fs)、持久性有机污染物(POPs)等新型污染物逐渐被国家所重视,如何实现NO、VOCs和汞多污染物协同控制已经成为目前研究的重点。彭悦[91-95]研究了HCl和氯苯共同作用下对MnCe和钒基催化剂的中毒影响,结果发现HCl对催化剂氧空穴攻击为催化剂失活的主要因素,氯苯在低温条件下不完全氧化产生的焦炭可以在SCR催化反应过程中被NO2进一步氧化为CO2和HCl。根据以上研究可知,NO2和HCl的存在可以极大地促进Hg0的氧化,使Cl离子以HgCl2的形式存在,减少催化剂中毒,所以NO、PCCD/Fs和Hg0协同催化可能是一种多污染高效协同脱除路径。如图7所示,未来催化剂在NO、PCCD/Fs和Hg0协同脱除方面应该构建不同的吸附反应区域,达到NO还原与PCCD/Fs和Hg0氧化协同发生,减少不同污染物的竞争吸附,促进污染物催化过程中间产物(-NO2)或最终产物(HCl)对Hg0氧化效果,最终达到多污染物协同脱除的效果。

图 7 Hg0等多污染物协同催化脱除技术反应机理[91-95]Fig. 7 Reaction mechanism of multi-pollutants synergy catalysis removal[91-95]
3 结论与展望
本文系统综述了Hg0催化氧化的最新研究进展,着重介绍了催化剂的研究现状和发展前景。将Hg0氧化催化剂分为钒基催化剂、贵金属催化剂和过渡金属氧化物催化剂,对催化剂的优缺点进行了详细的阐述和讨论。钒基催化剂汞氧化能力主要依赖于烟气中HCl和Cl2的浓度。贵金属催化剂在酸性气体中对Hg0有选择性吸附,有利于催化剂抗SO2和抗NH3。过渡金属氧化物在中低温下具有良好的Hg0氧化性能,但NH3和SO2严重抑制了催化剂活性。过渡金属氧化物催化剂在低温下的耐NH3和SO2性能是亟待解决的问题。
SO2和NH3与气态Hg0竞争活性位点,导致催化剂失活。从目前的研究可以看出,纯金属氧化物作为单一氧化位点并不适合烟气中汞的催化氧化。多种金属氧化物分别为SO2和Hg0提供更多的活性位点。催化剂表面的碱度增强是提高耐氨性能的可行途径。因此,在SO2和NH3存在下,通过多种金属组合和碱酸性质的调整,进一步提高Hg0氧化催化剂抗硫抗氨性。此外,在SO2和NH3存在下,贵金属(Au、Pt、Pd和Ag)对Hg0有很好的选择性吸附能力,贵金属可以吸附汞作为Hg0氧化的第一步。
现有的汞氧化催化剂反应温度窗一般在200 ℃以上。在目前的研究中,高效的低温汞氧化催化剂尚不多见。低温催化剂可以放置在除尘设备之后,从而降低NH3的负面影响,因此,低温汞氧化催化剂是一项重要的研究内容。低温催化剂可以吸附氧化Hg0生成HgO,但低温脱附HgO是一个关键问题。因此,未来研究方向应是降低脱附能和反应温度。
猜你喜欢
化工管理(2022年13期)2022-12-02
分子催化(2022年1期)2022-11-02
贵金属(2021年1期)2021-07-26
贵金属(2021年1期)2021-07-26
——庆祝中国共产党成立一百周年贵金属纪念币展
中国钱币(2021年4期)2021-02-26
陶瓷学报(2020年6期)2021-01-26
中学生数理化·中考版(2018年11期)2019-01-31
测控技术(2018年2期)2018-12-09
教学考试(高考化学)(2018年5期)2018-12-06
中国资源综合利用(2016年2期)2016-01-22
