
基于酵母同源重组的转录因子活性分析
2023-07-14 01:43李万通周逍灵
安徽农业大学学报 2023年3期
裴 浩,李万通,周逍灵,余 敏,陶 芳
基于酵母同源重组的转录因子活性分析
裴 浩,李万通,周逍灵,余 敏,陶 芳*
(安徽农业大学生命科学学院,合肥 230036)
转录因子的转录激活验证是分析其转录活性的有效手段之一。黄曲霉菌基因广泛参与该菌的生长发育,因此拟进行该转录因子的体外活性检测,以pGBKT7为骨架载体,采用酵母同源重组法构建载体,与体外重组酶法进行比较,并考察不同长度同源序列对酵母同源重组法构建载体的影响。结果表明:采用外源重组酶法构建载体pGBKT7-bZIP5,需要经过片段的体外重组连接、转化大肠杆菌,再提取质粒转化酵母AH109进行激活验证;而酵母同源重组法只需将线性化载体和片段一起转化酵母AH109,载体构建与激活验证同步完成,且21 ~ 30 bp不同长度的同源序列对酵母同源重组法构建载体没有影响,目的基因均显示有自激活能力,故基于酵母同源重组的载体构建方法可行。该方法无需提供外源重组酶,为基于酵母的载体构建提供了便捷的途径。
载体构建;同源重组;酵母;bZIP5转录因子
传统的酶切/酶连的载体构建的方法,是将目标载体、目的基因经限制性内切酶消化,T4DNA连接酶将其连接构成的重组载体,转化大肠杆菌感受态细胞后进行单克隆筛选。但是,这一方法受限于目标序列/载体的酶切位点、限制性内切酶的选择和酶切效果,尤其是对于需要在载体中插入多个基因片段时,构建载体的效率极低。利用重组融合PCR法构建载体,简化了多基因片段的连接[1-5],但多数还需依赖酶切/酶连,将连接好的片段插入到载体中,对于目标序列中无法回避的酶切位点时,往往利用引物设计点突变来消除酶切位点[6]。且这一方法对DNA polymerase的要求较高,重组片段较长时易出现序列错误。为此,人们开发出重组酶法构建载体,又称无缝克隆法载体构建,它利用同源重组的原理,通过设计带有载体同源序列的引物进行目的基因扩增,通过外源重组酶将其与线性化载体共同孵育,使有同源序列的目的基因和线性载体进行体外重组连接,然后转入感受态大肠杆菌,筛选阳性克隆。这种方法相对传统的酶切/酶连法较为简单,可以快速有效地获得目标重组载体,但有研究表明,其假阳性率较高[7],其次成本不低。
酵母细胞内具有高效的同源重组修复双链DNA断裂(double-strand breaks,DSB)的机制[8-9]。利用这一同源重组系统,可以实现载体构建,主要依赖于外源基因片段两端与载体上同源序列,在酵母细胞内实现重组,拼接成目标载体[10-11]。本文对黄曲霉家族bZIP5转录因子在酵母体内进行转录激活验证,利用酵母自身的同源重组系统构建载体,并与外源重组酶法进行比较。同时,还考察了不同长度的同源序列对酵母体内重组法的影响,为载体构建提供更为便捷的途径。
1 材料与方法
1.1 供试菌株及载体
酵母AH109和载体pGBKT7为安徽农业大学杨俊老师惠赠。大肠杆菌DH5α,黄曲霉菌NRRL3357为本实验室保藏的菌株。
1.2 培养基
YPD培养基:10 g·L-1Yeast extraction,20 g·L-1Peptone,20 g·L-1D-glucose,20 g·L-1Agar (液体培养基不加)。单缺培养基(SD/-Trp):6.7 g·L-1Yeast nitrogen base W/O amino acids,20 g·L-1D-glucose(需单独灭菌),1.19 g·L-1二缺粉末(-Leu/-Trp),1 mmol·L-1Leu,20 g·L-1Agar。三缺培养基(SD/-Ade/-His/-Trp):Yeast nitrogen base W/O amino acids 6.7 g,D-glucose 20 g·L-1(需单独灭菌),四缺粉末(SD/-Ade/-His/-Leu/-Trp)1.15 g·L-1,1 mmol·L-1Leu,20 g·L-1Agar。
1.3 主要试剂
10×LiAc:1 mol·L-1LiAc, pH 7.5;1×LiAc/40% PEG-4000/1×TE:1 mL 10×LiAc、1 mL 10×TE、4 g PEG-4000加水定容至10 mL,过滤除菌。
1.4 重组酶法载体构建
以黄曲霉NRRL 3357的cDNA为模板,-21F/-21R(表1)为引物对,TransStart Fast酶(北京,全式金)扩增转录因子基因CDS片段并测序。以RⅠ和HⅠ(Takara)双酶切载体pGBKT7,使之线性化。于基因片段和线性载体中加入重组酶Exnase II(诺唯赞,南京),获得pGBKT7-bZIP5重组载体,转化DH5α。以引物对-21F/-21R 和T7-f/BD-r(表1)分别进行PCR验证。
1.5 酵母感受态细胞的制备
挑AH109单菌落于5 mL YPD培养基中,30 ℃,230 r·min-1,振荡培养18 h。取菌液约1 mL于50 mL YPD液体培养基,使其OD600为 0.4,继续恒温振荡培养约5 h,至其 OD600为1.0左右。2 500 r·min-1,4 ℃,离心5 min;加入40 mL 1×TE后再次重悬并离心;最后加入2 mL 1×LiAc/0.5×TE,备用。
1.6 热激法转化酵母感受态细胞
取pGBKT7-bZIP5重组质粒5 μL,加入10 μL变性鲑精DNA(10 mg·mL-1)混匀。再加入100 μL感受态酵母细胞、700 μL 1×LiAc/40% PEG-4000/ 1×TE,于30 ℃、200 r·min-1条件下,振荡培养0.5 h。添加88 μL二甲亚砜并充分混合,42 ℃水浴5 min后,冰浴2 min。8 000 r·min-1离心10 s,留沉淀。加1×TE 100 μL重悬细胞。取50 μL涂布于单缺培养基和三缺培养基上,于30 ℃恒温培养,直至观察到菌落的出现。同时以酵母感受态细胞、转化了空载体pGBKT7的酵母细胞为对照。

表1 本研究所用的引物
1.7 酵母同源重组一步法构建载体
设计含不同长度同源序列的引物(表1),重组质粒pGBKT7-bZIP5作模板,PCR扩增目的基因片段。RⅠ和HⅠ双酶切载体pGBKT7。将线性载体pGBKT7和片段按1:2的摩尔比混合,转化酵母感受态细胞,涂布于单缺培养基和三缺培养基上。将三缺板上长出的单菌落,分别点涂到单缺和三缺培养基上面,再次筛选验证。挑取三缺板上的单菌落,提取质粒为模板,以不同长度同源序列的引物,用Easy酶(北京,全式金)进行菌落PCR。同时,以T7-f/BD-r为引物对,再次PCR测序验证。
2 结果与分析
2.1 重组酶法构建载体及bZIP5自激活探究
本研究利用酵母双杂载体pGBKT7进行转录因子bZIP5自激活验证,基因将插入到pGBKT7载体的多克隆位点(图1)。利用重组酶法,将bZIP5转录因子CDS序列(831 bp)与线性化的pGBKT7进行重组。按照重组酶试剂盒的说明,基因片段扩增采用的是21 bp链接头的引物对-21F/-21R。将重组反应体系转化大肠杆菌,提取质粒,酶切结果表明,基因片段已插入pGBKT7载体中(图2A)。为进一步验证外源基因片段插入的准确性,对获得的pGBKT7-bZIP5重组载体,利用T7-f/BD-r引物进行PCR扩增并测序。T7-f/BD-r引物为载体pGBKT7上的测序引物,引物间的原序列长度为299 bp,在RⅠ和HⅠ之间插入基因片段后,引物间序列长度约为1 130 bp(图2B)。测序结果表明,pGBKT7-bZIP5载体重组正确。

图1 pGBKT7载体的限制性图谱和多克隆位点(MCS)
Figure 1 Restriction map and multiple cloning site (MCS) of pGBKT7
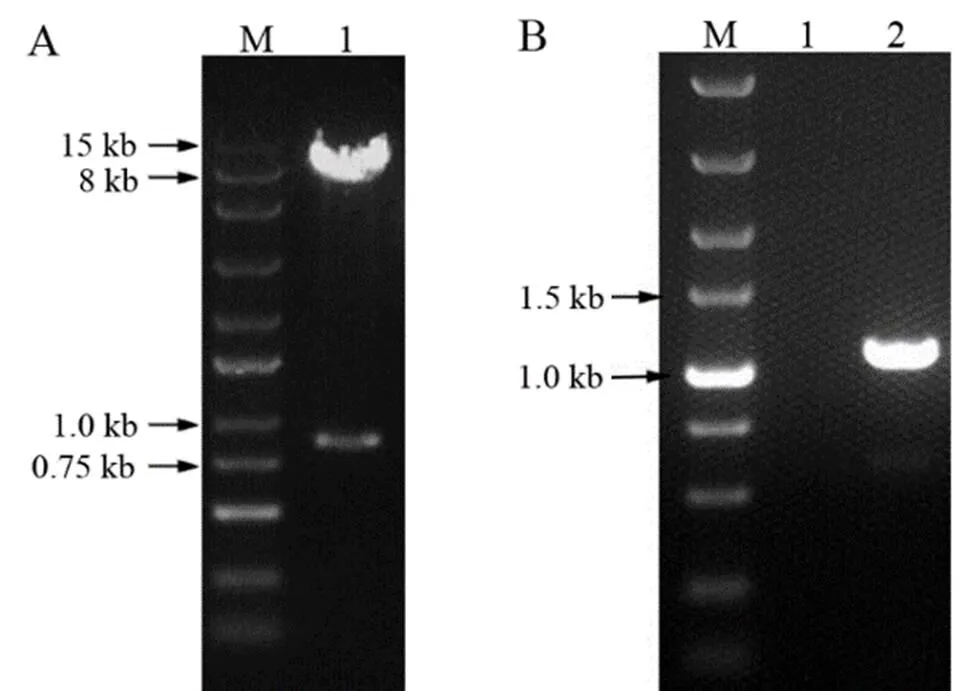
A.重组质粒酶切验证:M. 250 bp DNA Ladder; 1. EcoR Ⅰ/BamH Ⅰ酶切pGBKT7-bZIP5。B.重组质粒PCR验证:M. DL5000 Marker;1. 空白对照;2. pGBKT7-bZIP5为模板。
Figure 2 Verification of recombinant plasmid by recombinant enzyme method
将重组酶法构建的质粒pGBKT7-bZIP5转化酵母,分别涂布于单缺(SD/-Trp)和三缺(SD/-Ade/-His/-Trp)培养基上,同时以酵母感受态细胞AH109、转化了空载体pGBKT7的酵母细胞作为阴性对照。由于本实验所用酵母载体pGBKT7具有TRP1表达盒,故在单缺(SD/-Trp)培养基上,转化了空载体和重组载体的转化子均可生长。同时,载体pGBKT7具有BD结构域,若bZIP5是转录因子,则能激活酵母体内HIS3和ADE2这两种报告基因的表达,可启动Ade和His的合成,含有重组载体的细胞可在三缺(SD/-Ade/-His/-Trp)培养基上生长。基于此,转化结果显示,酵母感受态细胞AH109在单缺/三缺培养基上均未生长;转化了空载体pGBKT7的对照组,在单缺培养基上有菌落生长,在三缺培养基上无菌落出现;而转化了重组载体pGBKT7-bZIP5的酵母细胞在单缺和三缺培养基上均有菌落生长(图3)。将pGBKT7-bZIP5转化子点接于单缺和三缺培养基上,进一步验证了bZIP5是具有转录激活能力的转录因子(图4);提取转化子质粒,以T7-f/BD-r为引物对,PCR均扩增出目的条带(图5),表明为阳性转化子。
2.2 酵母同源重组法构建载体与bZIP5自激活验证
利用设计的3组不同长度同源序列的引物,扩增获得具有不同衔接头(21、25和30 bp)的片段,并通过酶切将pGBKT7载体线性化;将各片段分别与线性化载体一起转入酵母感受态细胞中,直接涂布于单缺和三缺培养基上。此法不同于上述重组酶反应体系的载体构建方法,无需重组酶,也无需转化大肠杆菌细胞。若酵母体内同源重组得到正确的重组载体,将会启动下游报告基因的表达,则三缺培养基上将出现转化子。以空载的pGBKT7载体为阴性对照,以重组酶法构建的pGBKT7-bZIP5为阳性对照。酵母重组法构建的载体分别命名为pGBKT7-21-bZIP5、pGBKT7-25-bZIP5和pGBKT7-30-bZIP5。结果显示,阴性对照在单缺培养基上有菌落生长,而三缺培养基平板上未观察到菌落生长;实验组和阳性对照组的结果一致,单缺培养基和三缺培养基平板上均有菌落生长(图6)。

图3 重组酶法构建的载体转化酵母细胞
Figure 3 Transformation of yeast cells with vector constructed by recombinant enzyme method

图4 pGBKT7-bZIP5转化子转录激活验证
Figure 4 Verification of transcriptional activation of pGBKT7- bZIP5 transformants

M: DL5000 Marker;1: 空白对照;2—4:pGBKT7-bZIP5转化子。
Figure 5 Verification of pGBKT7-bZIP5 transformants by PCR
将以上含不同衔接头重组载体的酵母细胞分别点接到单缺和三缺培养基上,以含空载体pGBKT7的酵母细胞为对照。结果显示,在单缺培养基平板上,含空载体pGBKT7的酵母细胞以及分别含重组载体pGBKT7-21-bZIP5、pGBKT7-25-bZIP5和pGBKT7-30-bZIP5的酵母细胞都能够生长。而在三缺培养基平板上,只有含重组载体的酵母细胞能够生长(图7)。

图6 酵母同源重组法构建载体
Figure 6 Construction of vector by yeast homologous recombination

图7 酵母同源重组转化子的转录激活验证
Figure 7 Verification of transcriptional activation of yeast homologous recombination transformants

M:DL5000 Marker;1—2:bZIP5 (bZIP5-21F/bZIP5-21R引物对);3—4:bZIP5 (bZIP5-25F/bZIP5-25R引物对);5—6:bZIP5 (bZIP5-30F/bZIP5-30R引物对)。
Figure 8 Verification of yeast homologous recombination transformants by colony PCR

M:DL5000 Marker;1,3和5:空白对照;2:重组载体pGBKT7-21-bZIP5;4:重组载体pGBKT7-25-bZIP5;6:重组载体pGBKT7-30-bZIP5。
Figure 9 Results of PCR amplification of yeast homologous recombinant plasmid
利用菌落PCR进一步验证酵母体内重组的载体,以3组不同长度衔接头的引物进行PCR,结果分析显示,实验组均扩增出约0.8 kb的目的基因条带(图8)。同时,以提取的重组质粒作为模板, T7-f/BD-r为引物对进行PCR扩增。结果表明,实验组均扩增出约1.1 kb的DNA条带,经测序验证正确,表明三缺培养基平板上长出的菌落所含载体重组正确(图9)。
综上所述,基于酵母载体pGBKT7序列所设计的3组不同长度衔接头的引物,对利用酵母同源重组法构建重组载体以及目的基因自激活的验证没有明显影响。上述结果表明,目的基因具有自激活能力,基于酵母体内同源重组进行载体构建的方法可行。
3 讨论
转录因子在真核生物的生长、发育、繁殖等生物学过程中发挥着重要作用。本实验室前期发现黄曲霉转录因子bZIP5参与黄曲霉毒素的合成以及菌核的发育[12],对该转录因子进行自激活验证,以pGBKT7为骨架载体,利用酵母体内的同源重组,成功构建了黄曲霉bZIP5转录因子激活验证载体,载体构建与转化子筛选一步完成,无需酶切/酶连,也无需加入外源重组酶,可直接进行菌落PCR验证载体构建是否正确,无需提取质粒,快捷方便,经济高效。而在重组酶法中,需将获得的重组载体先导入大肠杆菌扩繁,再提取载体质粒,导入酵母感受态细胞,步骤繁琐耗时。酵母同源重组法,只需将目的基因片段和线性化载体片段一起导入酵母感受态细胞培养,即可完成重组载体的构建和激活验证,减少了重组酶法导入大肠杆菌等操作所消耗的时间。
在利用酵母同源重组法进行载体构建时,本研究还考察了不同长度同源序列是否会影响酵母体内的同源重组,根据酵母载体pGBKT7的序列特征设计了3种不同长度(21 bp、25 bp和30 bp)衔接头的引物,3种长度同源序列均可实现重组。此外,我们还设计了16 bp衔接头,亦可获得重组载体(未发表)。也有研究表明,15 bp大小就可以实现重组克隆,但是重组率较低[13]。
该方法不仅可应用于酵母表达载体的构建,还可应用于其他非酵母表达载体构建,只要在载体中引入酵母质粒复制位点,并且在进行多个同源DNA片段重组拼接时,酵母同源重组会表现出更大的优势与潜力。Kilaru等曾使用基于酵母重组的克隆方法构建了pCeGFP载体以用于转化小麦叶枯病菌()[14];Lu等采用酵母重组克隆法构建了pKO1B敲除载体,对稻瘟菌()104个Zn2Cys6 转录因子进行了缺失功能分析[15];我们曾利用此法构建了黄曲霉菌双元表达载体,多达6个外源片段均可正确构建到载体中,通过农杆菌介导获得了目标菌株[12,16],说明酵母同源重组的高效与准确性。
[1] HOU X W, TONG H Y, HE Z H. Alternative seamless cloning strategies in fusing gene fragments based on overlap-PCR[J]. Mol Biotechnol, 2021, 63(3): 221-231.
[2] HILGARTH R S, LANIGAN T M. Optimization of overlap extension PCR for efficient transgene construction[J]. MethodsX, 2020, 7: 100759.
[3] 马彦, 岑雯, 刘昕, 等. 两步融合PCR法构建烟曲霉ssk1基因敲除株[J]. 山西医科大学学报, 2014, 45(4): 255-260.
[4] SZEWCZYK E, NAYAK T, OAKLEY C E, et al. Fusion PCR and gene targeting in[J]. Nat Protoc, 2006, 1(6): 3111-3120.
[5] JIANG X L, YANG J M, ZHANG H B, et al. In vitro assembly of multiple DNA fragments using successive hybridization[J]. PLoS One, 2012, 7(1): e30267.
[6] 邝翡婷, 袁定阳, 李莉, 等. 一种载体构建的新方法: 重组融合PCR法[J]. 基因组学与应用生物学, 2012, 31(6): 634-639.
[7] 马凯, 胡红霞, 于婧, 等. 双酶切和同源重组方法构建pMIR-reporter载体的比较[J]. 中国病原生物学杂志, 2015, 10(6): 495-499.
[8] SZOSTAK J W, ORR-WEAVER T L, ROTHSTEIN R J, et al. The double-strand-break repair model for recombination[J]. Cell, 1983, 33(1): 25-35.
[9] GAMBLE D, SHALTZ S, JINKS-ROBERTSON S. Recombinational repair of nuclease-generated mitotic double-strand breaks with different end structures in yeast[J]. G3-Genes|Genomes|Genetics, 2020, 10(10): 3821-3829.
[10] MIZUTANI K. High-throughput plasmid construction using homologous recombination in yeast: its mechanisms and application to protein production for X-ray crystallography[J]. Biosci Biotechnol Biochem, 2015, 79(1): 1-10.
[11] VAN LEEUWEN J, ANDREWS B, BOONE C, et al. Construction of multifragment plasmids by homologous recombination in yeast[J]. Cold Spring Harb Protoc, 2015, 2015(9): pdb.top084111.
[12] ZHAO Q Q, PEI H, ZHOU X L, et al. Systematic characterization of bZIP transcription factors required for development and aflatoxin generation by high-throughput gene knockout in[J]. J Fungi (Basel), 2022, 8(4): 356.
[13] MANIVASAKAM P, WEBER S C, MCELVER J, et al. Micro-homology mediated PCR targeting in[J]. Nucleic Acids Res, 1995, 23(14): 2799-2800.
[14] KILARU S, SCHUSTER M, LATZ M, et al. A gene locus for targeted ectopic gene integration in[J]. Fungal Genet Biol, 2015, 79: 118-124.
[15] LU J P, CAO H J, ZHANG L L, et al. Systematic analysis of Zn2Cys6 transcription factors required for development and pathogenicity by high-throughput gene knockout in the rice blast fungus[J]. PLoS Pathog, 2014, 10(10): e1004432.
[16] TAO F, ZHAO K, ZHAO Q Q, et al. A novel site-specific integration system for genetic modification of A[J]. G3-Genes|Genomes|Genetics, 2020, 10(2): 605-611.
Transcription factor activation analysis based on yeast homologous recombination
PEI Hao, LI Wantong, ZHOU Xiaolin, YU Min, TAO Fang
(School of Life Sciences, Anhui Agricultural University, Hefei 230036)
Transcriptional activation verification of transcription factor is one of the effective methods to analyze the activity. Theis involved in fungal growth and development in. To identify its transcriptional activity, expression vectors were constructed based on pGBKT7 through homologous recombination in yeast, and which were compared with the recombinant enzyme method. The results showed that the construction of pGBKT7-bZIP5 by exogenous recombinant enzyme was laborious because the recombinant vector should be transformed tofirst, and then the plasmid was extracted and transformed to yeast AH109 for activation verification. However, in the yeast homologous recombination method, the vector construction and activation verification were completed in one step by transforming the linearized vector and fragment into AH109 simultaneously. Furthermore, 21 - 30 bp of homologous sequences were sufficient for recombination, and thegene showed self-activation ability. Our results demonstrated that the method of vector construction based on yeast homologous recombination is feasible. This method provides a convenient way for the construction of yeast-based vector without providing exogenous recombinase.
vector construction; homologous recombination; yeast; bZIP5 transcription factor
10.13610/j.cnki.1672-352x.20230625.008
2023-06-26 16:17:55
Q786
A
1672-352X (2023)03-0544-06
2022-06-09
安徽省自然科学基金(2108085MC72)资助。
裴 浩,硕士。E-mail:19720367@stu.ahau.edu.cn
通信作者:陶 芳,博士,副教授。E-mail:taofang@ahau.edu.cn
[URL] https://kns.cnki.net/kcms2/detail/34.1162.S.20230625.1457.016.html
猜你喜欢
华人时刊(2023年1期)2023-03-14
汉字汉语研究(2021年2期)2021-08-30
汉字汉语研究(2019年2期)2019-08-27
中国调味品(2017年2期)2017-03-20
创新作文(小学版)(2016年16期)2016-11-11
现代检验医学杂志(2016年5期)2016-08-20
国外医药(抗生素分册)(2016年3期)2016-07-12
河北书画研究(2016年3期)2016-04-28
中国酿造(2016年12期)2016-03-01
中国粮油学报(2016年5期)2016-01-23
