
基于机器学习算法探讨甲状腺相关性眼病的免疫相关基因
2023-08-11 04:50戴玮然
广西医学 2023年10期
孙 悦 夏 宁 戴玮然
(1 重庆医科大学附属第一医院内分泌科,重庆市 400010; 2 广西医科大学第一附属医院老年内分泌科,广西南宁市 530021; 3 重庆医科大学附属第二医院心内科,重庆市 400010)
甲状腺相关性眼病(thyroid-associated ophthalmopathy,TAO)是一种以眼球后及眶周组织的浸润性病变为特征的特异性自身免疫性疾病。研究显示,25%~50%的Graves病患者会出现TAO[1],故TAO亦称为Graves眼病。TAO的主要临床特征包括眼球突出、泪腺增大、眼睑回缩、复视和暴露性角膜病变,严重时可发展为不可逆的视力丧失[2]。目前TAO的治疗手段主要包括基础治疗、药物治疗、放射治疗和手术治疗。其中,静脉使用糖皮质激素是中重度活动性TAO的一线治疗方案[3]。
有研究表明TAO 的病因及发病机制主要涉及环境、遗传和免疫等因素[4],其中免疫因素是TAO发生、发展的关键因素。TAO的病理学改变主要为炎症、脂肪形成和糖胺聚糖积累[5]。近年来,有学者认为眼眶成纤维细胞是免疫细胞浸润并释放促炎性细胞因子的主要靶点,也是疾病进展的积极参与者[6-7]。在TAO早期及后期,眼眶成纤维细胞被激活后可分泌多种细胞因子,如白细胞介素(interleukin,IL)-2、IL-4、IL- 5、IL-6、IL-10、γ-干扰素和肿瘤坏死因子α[5,8]。这些细胞因子促进单核细胞和巨噬细胞在眶周组织中增殖,并促使眼眶成纤维细胞分化,刺激富含透明质酸的基质积聚,进而引起免疫应答[9]。尽管目前关于TAO发病机制的研究日益增多,但其与免疫细胞之间的关系尚不明确,有待进一步研究。
近年来,基于高通量平台的微阵列技术已被广泛应用于在基因组水平上探索和识别疾病诊断及预后评估的潜在生物标志物。本研究通过生物信息学方法,从公共数据库下载TAO患者的基因表达矩阵,获取与TAO相关的差异表达基因(differentially expressed genes,DEGs),对DEGs进行富集分析,并利用机器学习算法进一步对这些基因进行筛选以获取特征性基因,分析特征性基因的诊断价值及与免疫细胞之间的相互作用,从而为TAO的诊断和治疗提供新思路。
1 资料与方法
1.1 数据下载及整理 从GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)下载TAO相关基因芯片数据集GSE185952和GSE58331,整理数据后共获得24例正常对照者(正常对照组)的样本和27例TAO患者(TAO组)的样本,样本均取材自人眶周组织。利用R 4.2.1软件的sva包对 GSE185952和GSE58331的数据进行批次校正及合并整理。
1.2 DEGs的获取 利用R 4.2.1软件的limma包对合并后的芯片数据进行差异分析,设置筛选条件为校正后P值<0.05和|log2FC|>1,获取DEGs,然后绘制热图及火山图。
1.3 富集分析 (1)利用R 4.2.1软件的clusterProfiler包对DEGs进行基因本体论(Gene Ontology,GO)功能富集分析和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析,其中GO功能富集分析项目包括生物过程、细胞组分和分子功能。设置筛选条件为P<0.05,将P值从小到大进行排序,取排名前10的条目进行展示。(2)利用R 4.2.1软件的dose包对DEGs进行疾病本体(Disease Ontology,DO)富集分析。(3)在MSigDB 数据库(https://www.gsea-msigdb.org/gsea/msigdb/)下载包含KEGG信息的基因集“c2.cp.kegg.v7.4.symbols.gmt”作为参考基因集,利用R 4.2.1软件的clusterProfiler包进行基因集富集分析(Gene Set Enrichment Analysis,GSEA),从而识别TAO组和正常对照组之间差异最显著的信号通路,以P<0.05为差异有统计学意义。
1.4 特征性基因的筛选 最小绝对值收敛和选择算子(least absolute shrinkage and selection operator,LASSO)算法是一种利用正则化方法来提高预测精度的回归分析算法,可借助R 4.2.1软件的glmnet包进行运算;支持向量机-递归特征消除(support vector machine-recursive feature elimination,SVM-RFE)算法中的支持向量机算法则是一种监督机器学习的技术,被广泛用于分类和回归分析[10],为了避免过拟合,可进一步采用递归特征消除算法从元数据队列中识别出诊断效能最高的基因集。故本研究采用LASSO算法和SVM-RFE算法两种机器学习算法,对DEGs进行运算分析,在LASSO算法中设置α=1,在SVM-RFE算法中设置sizes=c[2,4,6,8,seq(10,40,by=3)],筛选具有潜在诊断价值的特征性基因。最终将LASSO算法和SVM-RFE算法得到的基因取交集,获取具有潜在诊断价值的最佳特征性基因并绘制韦恩图。
1.5 特征性基因的诊断价值分析 为了检验最佳特征性基因的诊断价值,利用TAO组和正常对照组的样本数据,绘制特征性基因mRNA表达量诊断TAO的受试者工作特征(receiver operating characteristic,ROC)曲线,以曲线下面积(area under the curve,AUC)表示诊断价值,AUC越大表明诊断价值越高。
1.6 免疫细胞浸润分析 针对两组样本的基因表达数据,基于R 4.2.1软件通过CIBERSORT反卷积算法模拟计算22种免疫细胞的转录特征矩阵。利用R 4.2.1软件的corrplot包对各组内22种免疫细胞的占比进行可视化处理,再利用vioplot包绘制小提琴图,对TAO组和正常对照组之间22种免疫细胞占比的差异性(采用Mann-Whitney检验分析两组的差异性)进行可视化处理,然后利用pheatmap包绘制热图,展示22种免疫细胞的相关矩阵。
1.7 特征性基因与免疫细胞的相关性分析 利用R 4.2.1软件的Spearman秩相关检验,在TAO组中分析诊断价值相对较大的最佳特征性基因的mRNA表达量与免疫细胞占比之间的相关性,并使用ggplot2包对结果进行可视化展示。
2 结 果
2.1 DEGs的获取情况 共得到20个DEGs,其中表达上调基因7个,表达下调基因13个。上调基因分别为富含半胱氨酸/丝氨酸的核蛋白1(cysteine and serine rich nuclear protein 1,CSRNP1)、S100钙结合蛋白 (S100 calcium binding protein,S100)A8、早期生长应答蛋白1(early growth response 1,EGR1)、原癌基因C-Fos(proto-oncogene C-Fos,FOS)、G0/G1转换调节蛋白3 (G0/G1 switch regulatory protein 3,FOSB)、S100A12、核受体亚家族4 A组成员1(nuclear receptor subfamily 4 group A member 1,NR4A1 );下调基因分别为天冬酰胺内肽酶(legumain,LGMN)、巨噬细胞表达基因1(macrophage expressed gene 1,MPEG1)、软骨蛋白1(cartilage homeoprotein 1,ALX1)、凝血因子ⅩⅢ A1肽(coagulation factor ⅩⅢ A chain,F13A1)、跨膜结构域4亚家族A成员4(membrane-spanning 4-domains,subfamily A,member 4,MS4A4A)、差异表达蛋白2(differentially expressed protein 2,DAB2)、补体因子H(complement factor H,CFH)、锌指蛋白家族成员2(zinc finger protein family member 2,ZIC2)、可罗索β(Klotho beta,KLB)、淋巴管内皮透明质酸受体1(lymphatic vessel endothelial hyaluronan receptor 1,LYVE1)、半胱氨酸蛋白酶抑制剂A(cystatin A,CSTA)、基质重塑关联蛋白5(matrix remodeling associated 5,MXRA5)、甲状腺激素反应基因(thyroid hormone responsive,THRSP)。 见图1。
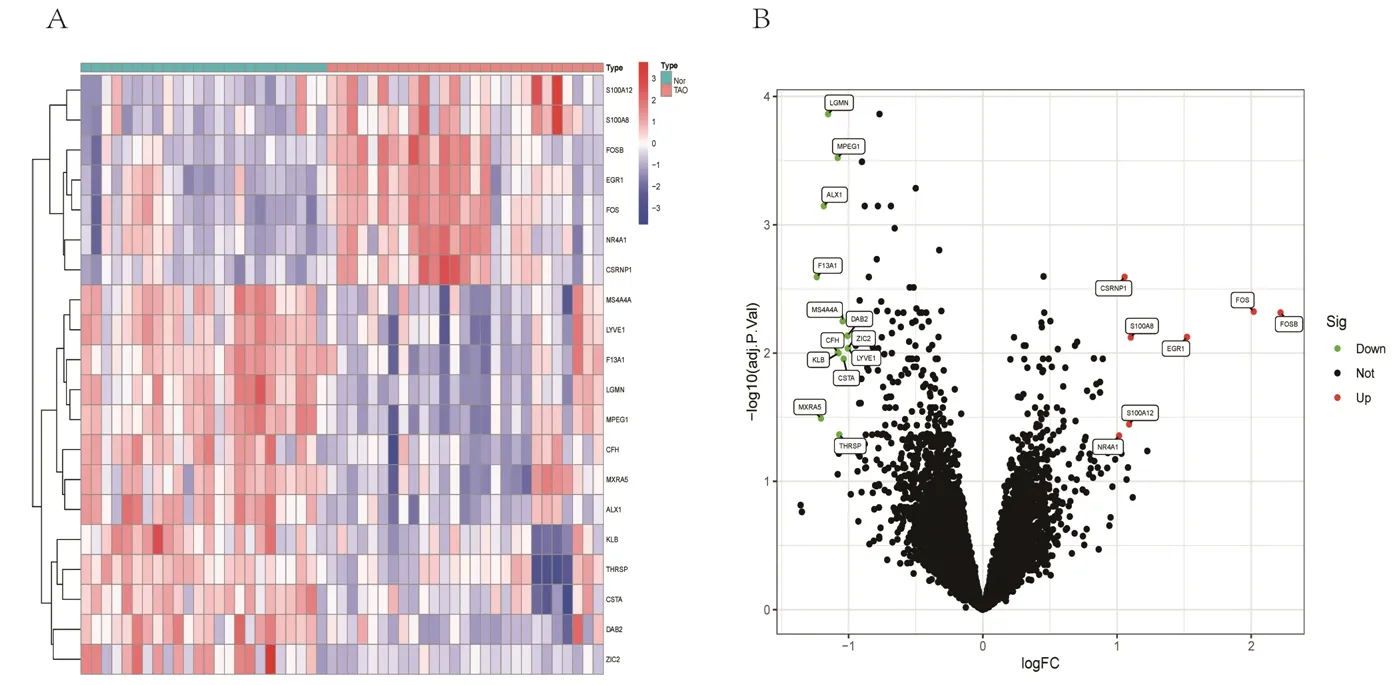
图1 DEGs的热图及火山图
2.2 富集分析结果 (1)GO功能富集分析结果显示,DEGs涉及的生物过程主要为骨骼肌细胞分化、激素代谢过程的正向调节、细胞趋化过程、烯烃化合物的生物合成、细胞对钙离子的反应等,涉及的细胞组分为分泌颗粒管腔及囊腔等,涉及的分子功能为DNA结合转录激活因子活性、多种受体(晚期糖基化终末产物受体、Toll样受体等)结合、维生素D依赖性钙结合蛋白、长链脂肪酸结合及氨基酰基转移酶活性等,见图2A。KEGG通路富集分析结果显示,大部分DEGs主要富集在IL-17信号通路、补体和凝血级联、甲状旁腺激素的合成与分泌、破骨细胞分化等信号通路中,见图2B。(2)DO富集分析结果显示,DEGs富集的疾病主要有食管肿瘤、关节炎、淋巴结疾病和川崎病等,见图3。(3) GSEA结果显示,与正常组相比,TAO组中的基因主要富集在与溶酶体、氧化磷酸化、氨基糖和核苷酸糖的代谢、N聚糖生物合成相关的信号通路及过氧化物酶体增殖物激活受体信号通路中,见图4。
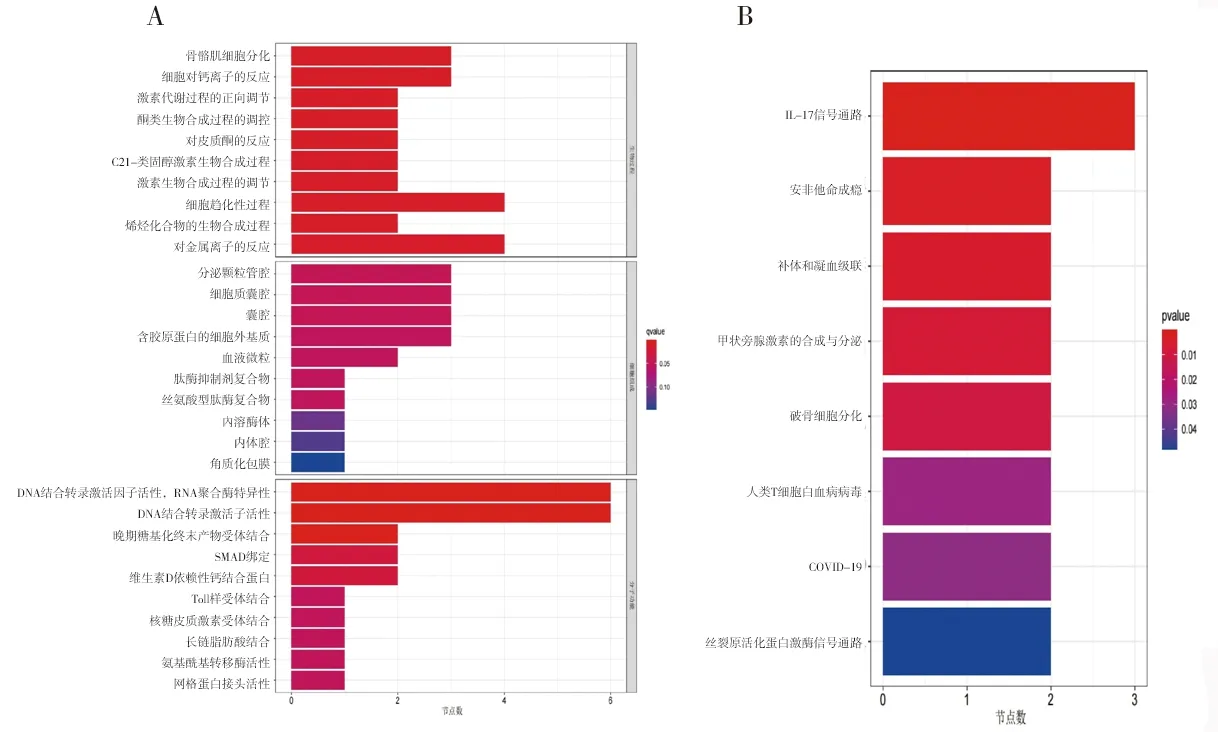
图2 DEGs的GO功能富集分析和KEGG通路富集分析结果
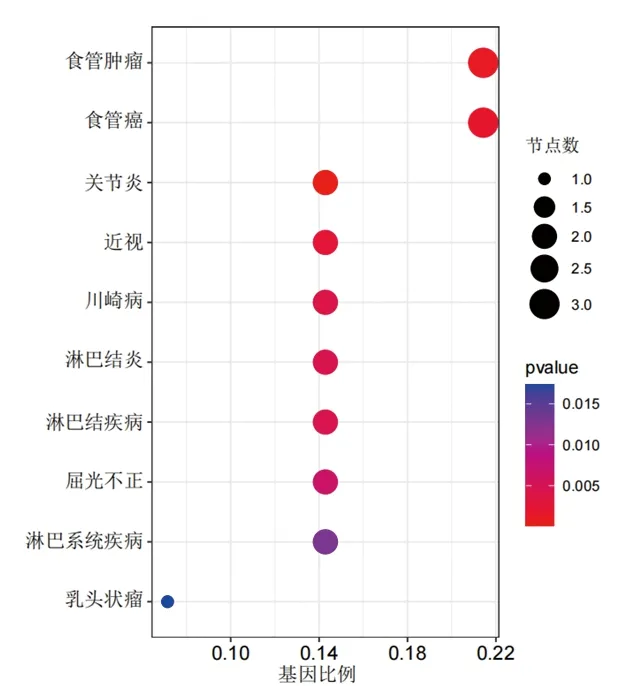
图3 DEGs的DO富集分析结果
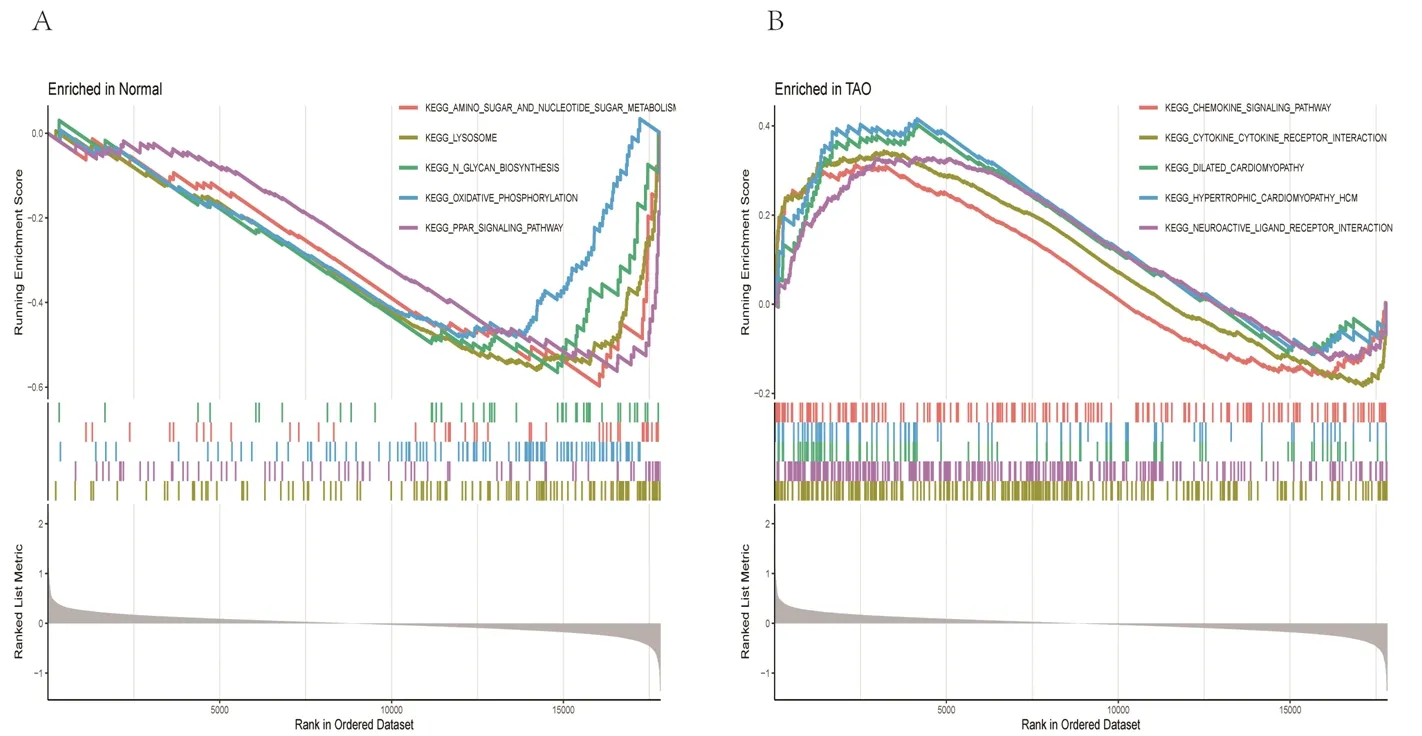
图4 GSEA结果
2.3 具有潜在诊断价值的最佳特征性基因的筛选结果 基于机器学习算法,对20个DEGs进行筛选。使用LASSO算法进行分析,得到ALX1、CSRNP1、KLB、MPEG1、S100A8、ZIC2 6个特征性基因,见图5A;使用SVM-REF算法进行分析,得到20个特征性基因,见图5B。对两种算法结果取交集后共获得6个最佳特征性基因,即ALX1、CSRNP1、KLB、MPEG1、S100A8、ZIC2,见图5C。
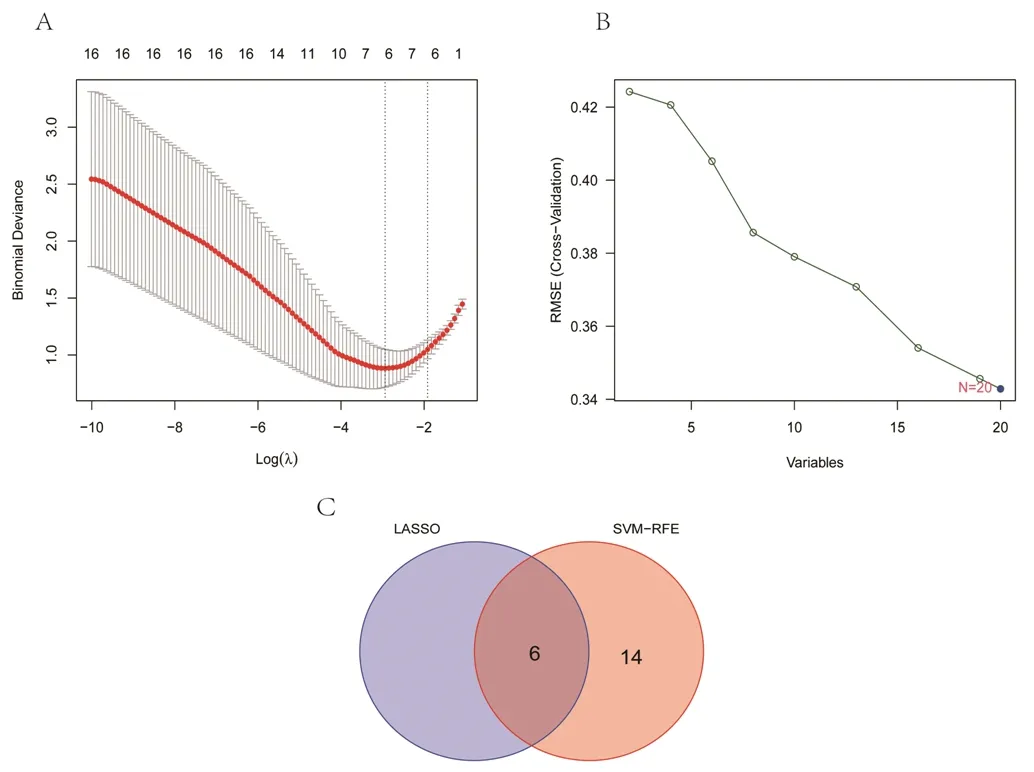
图5 基于机器学习算法筛选具有潜在诊断价值的最佳特征性基因
2.4 最佳特征性基因对TAO的诊断价值 ROC曲线分析结果显示,MPEG1的mRNA表达量诊断TAO的AUC相对较大,为0.889,提示其诊断价值较高,见图6。
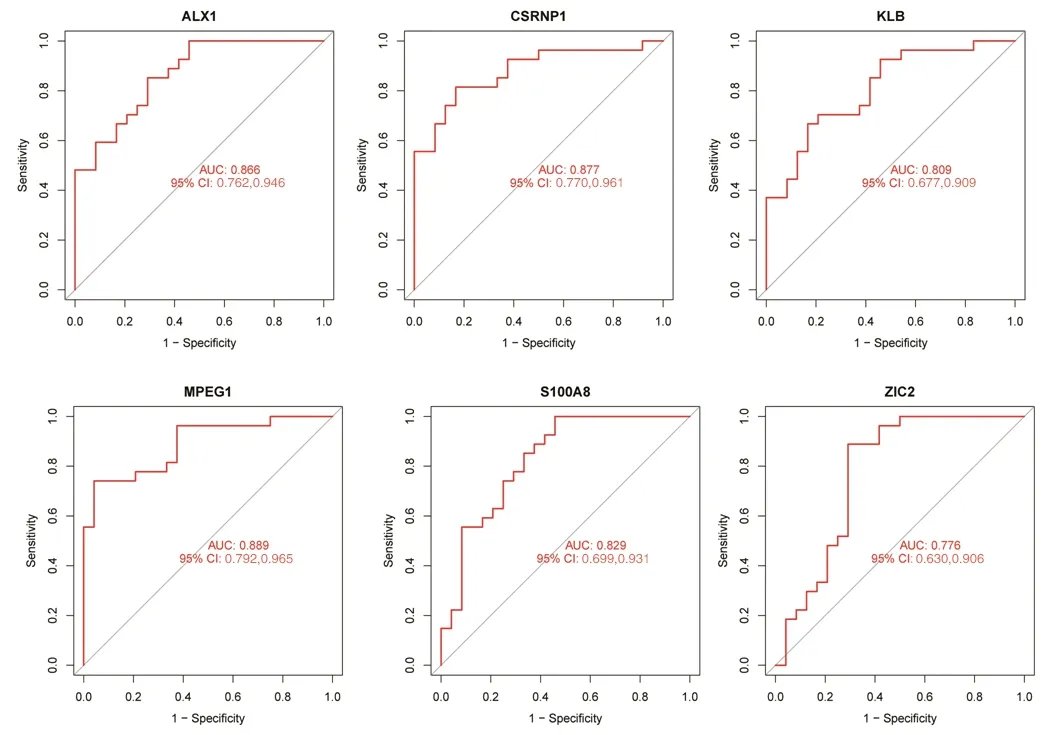
图6 6个最佳特征性基因mRNA表达量诊断TAO的ROC曲线图
2.5 免疫细胞浸润分析结果 正常对照组中,肥大细胞、树突状细胞及巨噬细胞的占比较大,而在TAO组中,中性粒细胞、嗜酸性粒细胞及自然杀伤(nature killer,NK)细胞的占比较大,见图7A。小提琴图显示,TAO组的CD4+幼稚T淋巴细胞、滤泡辅助性T淋巴细胞、单核细胞、M0型巨噬细胞、激活的肥大细胞及中性粒细胞的占比均高于正常对照组,而M2型巨噬细胞和静息的肥大细胞的占比均低于正常对照组(均P<0.05),见图7B。21种免疫细胞之间的相关性见图7C。
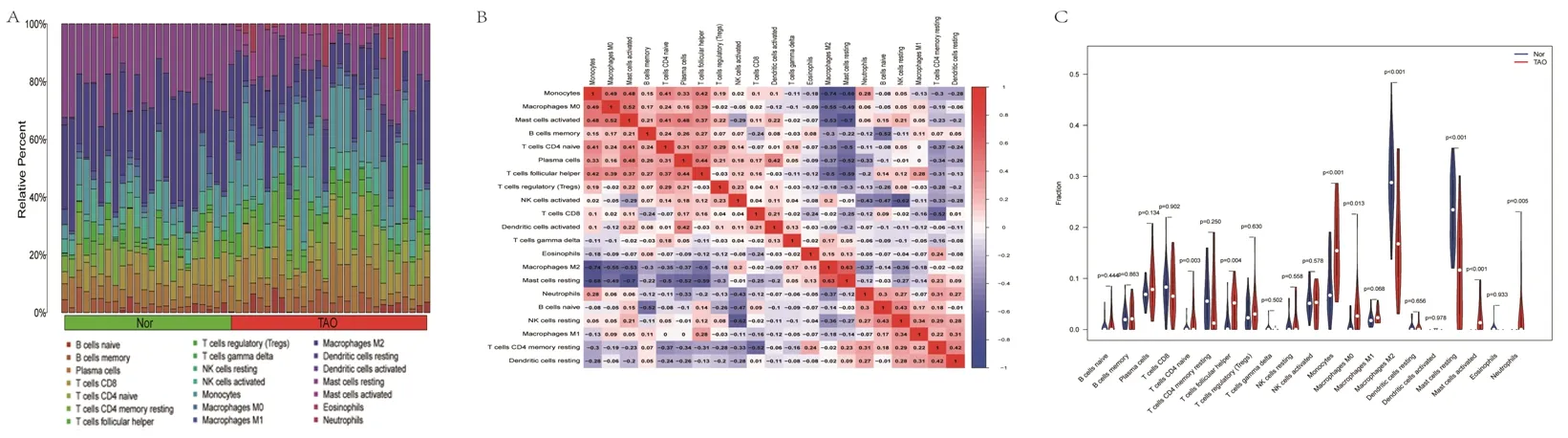
图7 免疫细胞浸润分析
2.6 在TAO组中特征性基因与免疫细胞的相关性分析 对2.4得到的诊断价值相对较高的最佳特征性基因MPEG1的mRNA表达量与免疫细胞占比进行相关性分析。结果显示,MPEG1的mRNA表达量与CD4+幼稚T淋巴细胞(r=-0.492,P<0.001)、滤泡辅助性T淋巴细胞(r=-0.780,P<0.001)、单核细胞(r=-0.544,P<0.001)、M0型巨噬细胞(r=-0.524,P<0.001)、激活的肥大细胞(r=-0.662,P<0.001)占比呈负相关,与M2型巨噬细胞(r=0.681,P<0.001)和静息的肥大细胞(r=0.730,P<0.001)占比呈正相关。见图8。
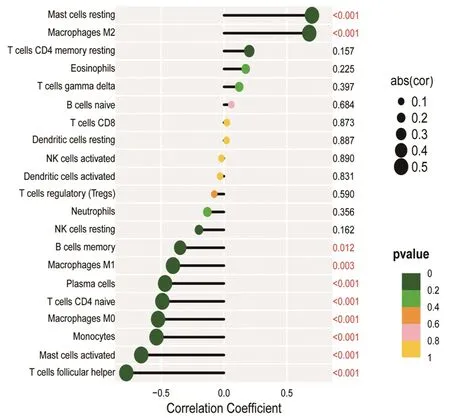
图8 TAO组中MPEG1的mRNA表达量与免疫细胞占比的相关性
3 讨 论
TAO是一种复杂的自身免疫性疾病,其确切的分子机制仍尚未完全明确[11],治疗手段仍相对有限。近年来,随着生物信息学的广泛应用,关于TAO发病机制及潜在靶点的研究日益增多。但是,由于TAO患者眶周组织标本获取困难,多数研究集中在患者血液标本的测序比对,准确性相对较差。此外,免疫细胞与生物靶点之间相关性的研究仍比较匮乏。因此,本研究利用机器学习算法探索TAO的免疫相关基因,以期为探寻TAO治疗靶点的相关研究提供参考依据。
本研究共获得20个与TAO相关的DEGs,GO功能富集分析结果显示,这些DEGs涉及的生物过程包括骨骼肌细胞分化、细胞趋化过程等,涉及的细胞组分包括分泌颗粒管腔及囊腔等,涉及的分子功能包括多种受体(晚期糖基化终末产物受体、Toll样受体等)结合、长链脂肪酸结合及氨基酰基转移酶活性等,说明DEGs主要与分子趋化、受体激活及炎症因子有关。研究表明,趋化因子和黏附分子可介导T淋巴细胞和B 淋巴细胞进入眼部组织,参与TAO的病理过程[12-13]。李威等[14]发现,在活动期TAO患者的泪液中IL-17、IL-10、IL-6 等炎症因子水平显著高于稳定期TAO患者,提示这些炎症因子对TAO的发生有重要作用。国外已有研究表明,在细胞因子过氧化物酶体增殖物激活受体γ作用下,TAO患者的眼外肌细胞分泌C-X-C基序趋化因子配体10和C-C基序趋化因子配体2增多,导致TAO症状加重[15]。王秋怡等[16]也发现,免疫细胞被激活后,趋化因子分泌增多,并可作用于眼眶成纤维细胞,引起眶周软组织炎症,使患者出现畏光、流泪、眼球突出等症状。研究显示,C-C基序趋化因子受体5不仅参与肿瘤的发生和发展过程,还在自身免疫性甲状腺疾病、糖尿病等许多自身免疫性疾病的发生和发展过程中有着重要的作用[17-18]。本研究的KEGG通路富集分析结果也显示,DEGs主要富集在IL-17信号通路上,说明IL-17信号通路与TAO密切相关,与既往研究结果[19]相符,IL-17有可能成为评估TAO疾病活动程度的新指标。DO富集分析结果显示,较多的DEGs在关节炎、淋巴结疾病和川崎病中丰度较高。GSEA结果也提示,与正常对照组相比,TAO组中的基因主要富集在与溶酶体、氧化磷酸化、氨基糖和核苷酸糖的代谢、N聚糖生物合成相关的信号通路及过氧化物酶体增殖物激活受体信号通路中。说明这些DEGs在免疫炎症性疾病的发生和发展过程中发挥重要作用,间接说明其可能也与TAO密切相关。
基于两种机器学习算法及ROC曲线分析,我们发现MPEG1的mRNA表达量对TAO的诊断效能较高。MPEG1是首个被发现的高表达于人类和鼠科动物巨噬细胞内的基因。有研究显示,在斑马鱼中转入MPEG1基因后其巨噬细胞系反应明显增强,说明MPEG1特异性表达于巨噬细胞[20]。MPEG1编码的蛋白为穿孔素2,但其在巨噬细胞防御机制中的作用尚不清楚[21]。近期还有学者在弥漫性大B细胞淋巴瘤中检测到MPEG1的突变,MPEG1的突变与T淋巴细胞激活和甲状腺、淋巴结等器官转移有着密切的关系[22]。然而,关于MPEG1与TAO之间相关性的研究仍相对匮乏,值得进一步探索。
国内已有研究证实TAO患者外周血中性粒细胞与淋巴细胞比值较正常人群升高,并与疾病的活动度相关[23]。本研究利用CIBERSOTR反卷积算法分析免疫细胞浸润情况与TAO的关系,结果也显示,CD4+幼稚T淋巴细胞、M0型巨噬细胞、中性粒细胞等多种免疫细胞在TAO中的占比高于正常组织。我们进一步在TAO组中进行相关性分析,结果提示MPEG1的mRNA表达量主要与单核细胞、巨噬细胞、CD4+T淋巴细胞等占比相关。近年来,国外学者通过对斑马鱼进行MPEG1蛋白荧光标记,并对其幼仔体内巨噬细胞和淋巴样祖细胞的转录组进行排序和测序,发现在巨噬细胞中可特异性检测到参与抗原呈递和加工的基因的表达,而在淋巴样祖细胞中可检测到参与巨噬细胞激活的基因的表达[24]。但是,以TAO患者为研究对象,探讨患者体内免疫细胞与MPEG1之间相互作用的研究仍处于空白。根据本研究的生物信息学分析结果,我们推测MPEG1极可能在TAO疾病的免疫应答中发挥关键作用。
综上所述,TAO是一种自身免疫性疾病,免疫细胞及炎症因子均参与该病的发生和发展。MPEG1可能是TAO的特征性基因,其与多种免疫细胞有着紧密联系,并与免疫细胞共同参与TAO的生理病理过程。这或可为研究TAO的发病机制及治疗药物提供新思路,但今后仍有待更深入的基础研究加以验证。
猜你喜欢
昆明医科大学学报(2022年4期)2022-05-23
中华养生保健(2020年4期)2020-11-16
广东农业科学(2017年5期)2017-08-29
哈尔滨医药(2016年3期)2016-12-01
癌变·畸变·突变(2016年3期)2016-02-27
现代检验医学杂志(2015年6期)2015-02-06
中医研究(2014年5期)2014-03-11
西南军医(2014年1期)2014-02-03
中国医学科学院学报(2013年2期)2013-03-11
食品科学(2013年19期)2013-03-11
