
2022年6、8月FDA批准新药概况
2023-08-22 01:32张建忠
上海医药 2023年15期
2022 年6 月,FDA 批出1 个新分子实体(表1),为治疗遗传性转甲状腺素蛋白介导的淀粉样变性药物Amvuttra(vutrisiran);8 月,FDA 批出1 个新生物制剂(表1),为治疗酸性鞘磷脂酶缺乏症药物Xenpozyme(olipudase alfa-rpcp)。
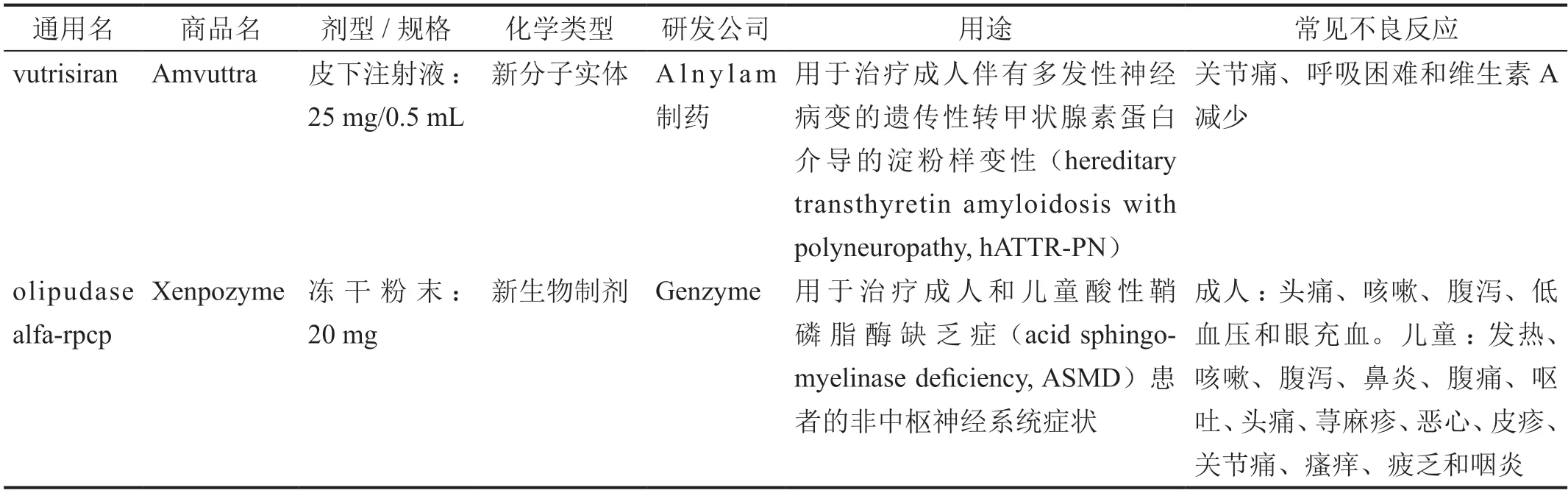
表1 2022年6、8月FDA批准新药
1 Amvuttra(vutrisiran)
Amvuttra 为注射液,被批准用于治疗成人hATTRPN——一种罕见的、遗传性的、快速进展的致命疾病,由转甲状腺素蛋白(transthyretin,TTR)基因突变引起。TTR 主要在肝脏中产生,正常情况下是维生素A 的载体。TTR 基因变异导致异常淀粉样蛋白蓄积,并损伤身体器官和组织,如外周神经和心脏,导致顽固性外周感觉-运动神经病变、自主神经病变和/或心肌病,以及其他疾病表现。患者诊断后的中位生存期仅为4.7 年,目前该领域存在重大的医疗需求未满足问题。Amvuttra 是一种双链小干扰RNA,靶向突变型和野生型TTR 信使RNA,阻断野生型和变体TTR 的产生。Amvuttra 基于Alnylam 制药公司的增强稳定化学(enhanced stabilization chemistry)型N-乙酰半乳糖胺(N-acetylgalactosamine)偶联递送平台设计,增加了效力和代谢稳定性,可允许不频繁的皮下注射。Amvuttra 的推荐剂量为25 mg,每3 个月皮下注射1 次,由医疗专业人员进行注射。
2 Xenpozyme(olipudase alfa-rpcp)
Xenpozyme 为冻干粉末,被批准用于治疗成人和儿童ASMD 患者的非中枢神经系统症状。ASMD 是一种极其罕见的遗传性疾病,也称为尼曼-匹克病,是由于缺乏分解鞘磷脂所需的酶引起的,该脂质在肝脏、脾脏、肺和大脑中积聚。ASMD 患者腹部增大,可引起疼痛、呕吐、喂养困难和跌倒,他们的肝脏和血液检查也存在异常。受影响最严重的患者有严重的神经系统症状,很少能活过两到三岁;其他患者可能会存活到成年,但会因呼吸衰竭而过早死亡。Xenpozyme 是一种水解溶酶体鞘磷脂特异性酶替代疗法用药,用于替代缺失或有缺陷的酸性鞘磷脂酶。该药物可降解鞘磷脂,进而帮助减少该脂质于肝脏、脾脏与肺中的累积。Xenpozyme 是第一个获批的用于治疗ASMD 患者与中枢神经系统无关的症状的药物。该药的儿童推荐起始剂量为0.03 mg/kg,每过2 周的剂量按0.1 mg/kg、0.3 mg/kg、0.3 mg/kg、0.6 mg/kg、0.6 mg/kg、1 mg/kg、2 mg/kg、3 mg/kg 的顺序使用;成人推荐起始剂量为0.1 mg/kg,每过2 周的剂量按0.3 mg/kg、0.3 mg/kg、0.6 mg/kg、0.6 mg/kg、1 mg/kg、2 mg/kg、3 mg/kg 的顺序使用。儿童和成人推荐维持剂量都是3 mg/kg,静脉输注。Xenpozyme 的标签附带一个黑框警告,其有包括过敏反应在内的严重超敏反应的风险。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
昆明医科大学学报(2022年4期)2022-05-23
基层中医药(2021年8期)2021-11-02
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
临床医药文献杂志(电子版)(2017年11期)2017-05-17
当代医药论丛(2017年22期)2017-04-12
中国卫生标准管理(2015年15期)2016-01-15
西南军医(2015年1期)2015-01-22
