
基于模糊支持向量机的赖氨酸糖化位点预测
2023-09-14 13:29宋一明张万里
沈阳航空航天大学学报 2023年3期
宋一明,鞠 哲,张万里
(沈阳航空航天大学,理学院,沈阳 110136)
作为一种常见而重要的蛋白质的翻译后修饰(Post-Transational Modifications,PTMs),赖氨酸糖化可以潜在地影响多种生物过程,如构象、功效和免疫原性等[1]。糖化是糖分子(如果糖或葡萄糖)与蛋白质或脂质分子共价结合的典型过程。与糖基化需要酶的控制作用相比,糖化是一种非酶修饰的过程。首先,不稳定的席夫碱(Schiffbase)重新排列,形成更稳定的阿马多里产物(n-substituted 1-amino-1-de‐oxy-ketose),随后,阿马多里产物可进一步反应形成晚期糖化终产物(Advanced Glycation Endproducts,AGEs),该终产物为不可逆交联产物[2-3]。赖氨酸糖化可以发生在细胞内和细胞外的蛋白质中[4-5]。一般来说,细胞内的糖化比细胞外的糖化更为复杂,因为细胞质中的多种潜在来源也可以反应形成AGEs。糖化反应的动力学分析指出,稳定状态下的糖化量与葡萄糖浓度、蛋白质半衰期和糖化率成正比[6]。大量研究表明糖化与多种人类疾病的发生和发展密切相关,如糖尿病及其血管并发症、肾功能衰竭、帕金森病和阿尔茨海默病[7-9]。因此,破译糖化的分子机制和生物学功能对上述疾病的治疗具有重要意义。
目前,糖化的分子机制在很大程度上仍是未知的。为了更好地理解糖化的分子机制,需要高精度地识别糖化底物及其相应的糖化位点。大规模蛋白质组学方法如质谱分析,已被用于检测糖化位点[10-11]。然而传统的实验方法不仅花费高,并且耗时耗力,很大程度上延缓了相关研究的进展。因此,有关蛋白质糖化的计算辅助方法受到了越来越多的关注。到目前为止,已有很多学者通过机器学习算法对糖化位点进行了预测。Johansen等[12]利用人工神经网络算法提出了第一个预测赖氨酸糖化位点的预测器NetGlycate,最终得到的马氏相关系数为0.77,AUC 值为0.58,体现了使用机器学习算法进行蛋白质糖化位点预测的可行性。Liu 等[13]提出了一种预测糖化位点的计算方法PreGly,该方法使用氨基酸因子、氨基酸出现频率和k 间距氨基酸对组成进行特征提取,使用最大相关和最小冗余(mRMR,max Relevance and Min Redundancy)进行特征选择,在k =4 时获得了最优的模型。Xu 等[14]开发了一种名为Gly-PseAAC 的预测器,利用位置特异性氨基酸偏好提取蛋白质包含的信息,然后使用支持向量机(Support vector Machine,SVM)算法预测糖化位点,通过PSAAP特征有效地验证了赖氨酸是否发生糖化反应的问题。Ju 等[15]提出了BPB(Bi-Profile Bayes)的特征提取方式,并结合支持向量机进行预测,预测的结果要优于以上算法。
然而,标准的SVM算法会因数据中存在野点或噪声而导致分类精度下降,因此Lin 等[16]提出了模糊支持向量机(Fuzzy Support Vector Machine,FSVM)方法,其思想为给每个样本以不同的隶属度,可以有效地降低野点或噪声对分类精度的影响。在此基础上,学者们提出了更多的隶属度函数设计方法,如文献[17]将样本的不确定性和样本与类中心的距离相结合,提出了一种基于信息熵的改进FSVM 算法,在不平衡数据集上具有较高的分类精度。李村合等[18]通过加入参数来调整分离超平面与样本的距离,当样本分布不均时也能得到较高的分类精度。Wang 等[19]提出了基于中心核对齐的模糊支持向量机。左喻灏等[20]提出了Re‐lief-F 特征加权的FSVM 算法,通过赋予样本权重与特征权重来提高分类效率。本文在文献[15]的基础上,提出了一种基于两步特征加权的模糊支持向量机算法。首先,利用信息增益算法获取样本的特征权重;然后,选择信息增益最大的特征,计算其与剩余特征的斯皮尔曼相关系数,将最大的特征权重与其他特征的相关系数相乘并加到其他特征原有的权重上,得到新的特征权重;最后,将得到的特征权重应用到隶属度函数距离的计算与核函数的构建中,同时考虑样本的亲和度,通过样本内部的分布情况对隶属度函数做出进一步修正。本文将上述算法与BPB 的特征提取方式结合,提出了一种预测赖氨酸糖化位点的方法FS‐VM_GlySite,并用十折交叉验证,结果表明,FSVM_GlySite 的预测结果要优于现有的几种常用的预测模型。
1 材料与方法
1.1 数据集
本文使用文献[15]中的训练集对模型进行训练和测试。此训练集来自蛋白质赖氨酸修饰数据库CPLM[21],包含了223 个实验标注的糖化赖氨酸位点和446 个非糖化赖氨酸位点。使用滑动窗口表示数据集的赖氨酸残基K,与文献[15]设置相同,窗口大小设置为15,每个训练样本都表示赖氨酸残基K 下游和上游各有的7个残基的肽段。为了统一每个肽段的长度,添加虚拟残基“X”来填补没有足够残基的位置。这里将糖化多肽作为正类训练样本,而非糖化多肽作为负类训练样本。
1.2 Bi-Profile Bayes特征提取
给定序列片段S=s1s2...sn,其中sj(j=1,2,...,n)为单个氨基酸,n 为序列片段长度。S属于类C1或类C2,其中C1和C2分别表示糖化位点和非糖化位点。根据贝叶斯准则,假设sj(j=1,2,...,n)相互独立,则两类S 的后验概率可表示为
式(1)和式(2)可重新表示为
假设类别的先验分布是均匀的,即P(c1)=P(c-1),则决策函数可表示为
根据文献[22],式(5)可以进一步写成
1.3 基于两步特征加权的模糊支持向量机算法
支持向量机是一种流行的机器学习算法,被广泛应用于各种PTMs 位点的预测[23-25]。模糊支持向量机则是给每个样本以不同的隶属度,降低野点和噪声对分类的干扰。一个训练1,-1}为训练样本的标签,+1 代表正类,-1 为负类,si∈[0,1]为模糊隶属度,表示样本xi属于类yi的权重。FSVM模型为
通过求解上述问题得到最终的分类决策函数为
式中:K(xi,xj)为核函数,目的是将样本通过非线性映射ϕ(x),使其映入高维核空间。
设计出好的隶属度函数是模糊支持向量机的关键。本文使用的算法首先对特征进行两步加权,再将得到的特征权重应用到隶属度函数的设计与核函数的构建中,最后通过样本亲和度对隶属度函数作出修正,从而得到每个样本的隶属度。
1.3.1 进行特征加权
(1)计算出所有特征的信息增益Gain(k),如式(9)、(10)所示
式中:D 为数据集;|D|为数据集中的样本个数;D 中有h个类别标签Ki(i=1,2,...,h);|Ki,D|为D中标签为Ki的样本个数。若特征A 有v个取值Aj(j=1,2,...,v),|Dj|为D 中在特征A 上取值为Aj的集合。式(10)表示特征对样本集合不确定性的减少程度,为信息熵与条件熵之差。
(2)计算出特征之间的斯皮尔曼相关系数矩阵corr(i,k),如式(11)所示
式中:Ri和Si表示样本i取值的等级;-R和-S表示变量R和S的平均等级;N为样本个数。斯皮尔曼相关系数用来衡量两个变量之间的相关性大小,越趋近于0,两个变量之间的相关性越低,绝对值大于0.4则认定为具有一定相关性。
(3)找到信息增益值最大的特征,位置记为M,然后根据其与剩余特征的相关系数,找到相关系数大于0.4 的特征,以式(12)赋予最终的特征权重w(k),相关系数小于0.4 的不作处理。将已得到w(k)的特征忽略,对未赋予w(k)的特征重复上述过程,直至全部特征都被赋予新的w(k)
(4)对RBF 核函数K(xi,xj)=exp(-γ||xixj||2)作出修改,根据特征权重w 得到特征矩阵的对角矩阵形式
新的特征加权核函数为
1.3.2 设计模糊隶属度函数
(1)本文在距离计算上均使用特征加权距离方法,如式(13)所示
式中:l表示特征的个数。
(2)通过模糊C 均值算法得到样本的正负类中心x+cen、x-cen,计算dcen+i= d(xi,x+cen)、dcen-i=d(xi,x-cen),赋予特征加权隶属度s1(xi),如式(14)所示
模糊C 均值算法具体思路为:假定对数据集S 进行分类,样本xi属于第j个聚类中心cj的隶属度为μij,表达式如式(15)、(16)所示
式中:m 为隶属度因子;N 与H 分别表示样本个数与聚类中心数;||xi-cj||2表示xi到中心点cj的距离。要求最小化目标函数J,给定任意初值后进行迭代,当maxij{|μ(k+1)ij-μ(k)ij|}<ε 时停止迭代,表示继续迭代后μij已无明显变化;k为迭代次数;ε为误差阈值。
(3)计算样本的亲和度A(xi),如式(17)~(19)所示
式中:U(xi,D)、T(xi,D)分别表示删除每个样本前后样本间距离标准差的变化比率、样本均值的变化比率。当样本分散度低、紧密度高时,样本对数据集的影响就越大,样本的亲和度就越小[26]。
(4)将样本亲和度归一化,以此保证亲和度与s1(xi)在同等数量级上,得到隶属度s2(xi)=-A(xi),计算得到最终的隶属度函数s(xi)=s1(xi)+s2(xi)。再将s(xi)归一化,防止隶属度为负的情况的出现。
参数c的区间为c={2-5,2-4,...,215},参数γ的区间为γ={2-15,2-14,...,23}。为了防止数据集正负样本不平衡对分类精度的影响,本文在参数c 的设定上使用文献[27]的方式,对不同类样本赋以不同的惩罚项c+=c-(N-p)/p,其中c+、c-分别为少类样本与多类样本的惩罚项;p表示少类样本个数;N-p为多数类样本个数。
1.4 交叉验证和性能评估
本文使用十折交叉验证来评估模型,具体为灵敏度(SN)、特异度(SP)、准确率(ACC)、马氏相关系数(MCC)和ROC 曲线下面积(AUC)5个指标,前4个指标定义为
式中:N+为糖化位点个数;N+-为错误预测为非糖化位点的糖化位点个数;N-为非糖化位点个数;N-+为被错误预测为糖化位点的非糖化位点个数。
2 结果和讨论
2.1 FSVM_GlySite的表现
通过十折交叉验证得到的AUC 值作为模型FSVM_GlySite 的评价指标。 由于FS‐VM_GlySite 是在文献[15]的训练数据集上进行训练的,本文还将FSVM_GlySite与BPB_Gl‐ySite[15]进行了比较。如表1 所示,FSVM_Gly‐Site 得到的SN、SP、ACC、MCC 和AUC(64.62%、73.92%、70.82%、37.27%和76.40%)均高于BPB_GlySite 方法(63.68%、72.60%、69.63%、34.99%和76.22%)。这是由于本文进行两步特征加权的方式可以最大程度地放大重要和次重要特征与弱相关和不相关特征在权重上的差值,有效地避免了后者对分类的干扰,加强了相对重要特征对分类的贡献,并且在考虑了样本亲和度后,衡量了每个样本的存在与否对数据集的影响;利用样本内部的分布情况对隶属度函数做出了适当修正,减少了仅使用样本与类中心距离作为隶属度函数时对数据集几何形状的依赖,降低了噪声和野点对分离超平面的干扰。本文使用了聚类的方式获得类中心,相比于求平均值计算出的类中心,其含有数据集中更多的样本信息,有助于获取更准确的样本隶属度值。虽然FSVM_Gl‐ySite 在SP 上低于Gly-PseAAC[14]的74.30%,但是在SN 上有着显著的提升(57.48%),说明FSVM_GlySite 可以更精确地识别赖氨酸糖化位点。
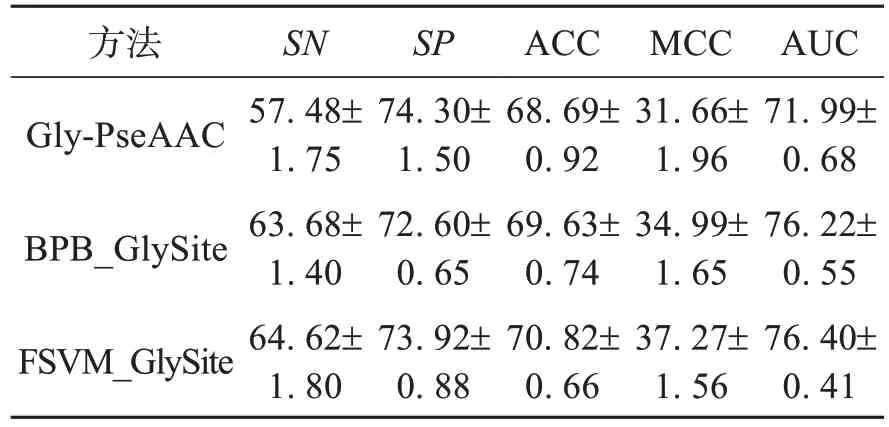
表1 训练集下的比较结果 (%)
2.2 FSVM_GlySite与现有预测方法的比较
为了进一步评估FSVM_GlySite 的有效性,将其与其他现有的预测方法进行比较,包括NetGlycate[12]、PreGly[13]、Gly-PseAAC[14]和BPB_GlySite[15]。由于NetGlycate 和PreGly 都是在包含89 个糖化位点和126 个非糖化位点的Johansen 基准数据集[12]上训练的。因此,将本文算法与BPB_GlySite 和Gly-PseAAC 也通过Johansen基准数据集上的十折交叉验证进行重新训练。比较结果见表2,其中FSVM_Gl‐ySite 的SN、ACC、MCC 和AUC 值最高,分别为87.64%、87.91%、75%和92%。虽然PreGly 获得了最高的SP值(95.85%),但Sn值(71.06%)远低于FSVM_GlySite(87.64%),这表明PreG‐ly 倾向于将赖氨酸残基预测为非糖化位点,其识别的糖化位点明显少于FSVM_GlySite,因此在Johansen基准数据集上,FSVM_GlySite同样优于现有的糖化位点预测器。
3 结论
本文提出一种新的基于两步特征加权的模糊支持向量机算法,并结合Bi-Profile Bayes方法构建了一个赖氨酸糖化位点预测模型FS‐VM_GlySite。实验结果表明,模糊支持向量机算法的分类性能效果好于标准的支持向量机算法,并且所提出的预测模型的预测效果优于现有的糖化位点预测方法。然而,本文所采用的是单一的特征编码方法,后续的工作将会尝试融合更多特征的方法,进一步提升预测模型的性能。
猜你喜欢
小学生学习指导(低年级)(2021年9期)2021-10-14
中学生数理化·七年级数学人教版(2019年10期)2019-11-25
小学生学习指导(低年级)(2019年9期)2019-09-25
小学生学习指导(低年级)(2018年9期)2018-09-26
酒·饮料技术装备(2018年1期)2018-04-28
广东饲料(2016年3期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
中国卫生标准管理(2015年25期)2016-01-14
动物营养学报(2015年10期)2015-12-01
应用化工(2014年10期)2014-08-16
