
电-酶偶联催化转化CO2的研究进展
2023-10-10 06:22吕永琴苏海佳秦培勇陈必强谭天伟
生物加工过程 2023年5期
吕永琴,苏海佳,秦培勇,陈必强,谭天伟
(北京化工大学生命科学与技术学院 北京市生物加工过程重点实验室 国家能源生物炼制研发中心,北京 100029)
化石燃料的过度利用导致人为CO2大量排放。每年释放的CO2超过350亿t,约为自然环境吸收速率的2倍[1-2]。预计到21世纪末,大量排放的CO2将导致全球地表温度升高约2 ℃[3]。根据2015年签订的《巴黎协定》,世界上大多数国家同意采用不同方式进行能源管理,以减少CO2的排放[4-8]。我国在2020年提出了2030年实现“碳达峰”,2060年实现“碳中和”的“双碳”战略目标。同时,CO2也是储量丰富的可再生碳源,可用于合成含碳燃料、材料和化学品。因此,实现对CO2的高效资源化利用是保障经济高质量发展、助力实现全球“双碳”目标的一种有效策略。
CO2的催化转化存在多种方法,主要包括热催化加氢[9-10]、光催化转化[11-12]、电催化转化[13]和生物催化转化[14]等(表1)。其中,电催化CO2还原具有反应条件温和、反应速率快和便于大规模工业应用等独特优势而备受关注,被视为实现碳中和最有前景的策略之一[15-16]。然而,电催化CO2还原的产物多局限于一碳化合物,如CO、CH4、甲酸和甲醇等,并且产物的选择性较低。生物催化转化CO2是利用活细胞或酶作为催化剂,从CO2合成复杂有机分子的过程。与化学催化剂相比,生物酶催化剂具有选择性高、操作条件绿色温和以及易实现碳-碳偶联、碳链延长等优点,因此有助于将CO2转化为高附加值长碳链产品[17]。根据目标产物的不同,可以选择利用单一酶或多酶作为催化剂。相对于单一酶催化剂,多酶催化剂系统对于生产高附加值产品更具优势。但由于CO2的高化学惰性,C=O键(750 kJ/mol)的断裂需要输入大量的能量,这使得CO2的活化和断键成为一个难题。同时,CO2在液相反应体系中的低溶解度严重影响了酶对底物的亲和力和催化效率。

表1 用于CO2转化的催化方法及各自特点
基于此,研究人员提出了电-酶偶联催化转化CO2策略,即在生物电化学体系中,利用电能驱动酶催化C=O断键,进一步实现碳-碳偶联和碳链的延长,以高能量效率合成高附加值和长碳链化合物。电-酶偶联催化为转化CO2生产附加值燃料、化学品和材料提供了一种新方法。因此,对这一新兴领域的最新研究进展及挑战进行系统性综述是必要的。本文综述了酶促电催化CO2还原的最新研究进展,详细介绍了NAD(P)H非依赖型和NAD(P)H依赖型氧化还原酶的电子传递机制,并重点阐述了酶与电极间的相互作用、电极的修饰与改性和新型电-酶耦合体系的构建及研究动态。此外,对电-酶偶联用于CO2催化转化所面临的主要挑战和未来发展前景进行了概述。
1 电子转移机制
由于各类酶的催化机制不同,电-酶偶联系统可以分为两类:NAD(P)H依赖型和NAD(P)H非依赖型系统(图1)。在NAD(P)H依赖型系统中,氧化型辅因子NAD(P)+接收电子再生为NAD(P)H,NAD(P)H作为电子和质子的载体,辅助酶催化CO2还原,并重新转变为氧化态的NAD(P)+。在NAD(P)H非依赖型的系统中,氧化还原酶可以通过直接电子转移(direct electron transfer,DET)或间接电子转移(mediated electron transfer,MET)两种方式接受来自阴极的电子。

图1 电-酶偶联催化CO2转化的电子转移机制Fig.1 Mechanism of electron transfer in electrocatalytic-enzymatic hybrid systems for CO2 reduction
1.1 NAD(P)H依赖型系统
NAD(P)H是还原型辅因子烟酰胺二嘌呤核苷酸及其磷酸化形式的简称。在NAD(P)H依赖型的电-酶偶联系统中,实现CO2还原通常需要经历多个电子转移步骤,其中辅酶NAD(P)+向NAD(P)H的转化是酶促CO2还原反应发生的先决条件。通过向阴极施加一定的电压(大于-0.32 VvsNHE[18]),氧化型的NAD(P)+可以从阴极上获取电子,反应所需的质子则来源于阳极上发生的氧化反应,通常为析氧反应。NAD(P)H在金属电极上直接催化再生的过电位较高,容易生成无活性的副产物[19]。为了解决这一问题,通常需要向电解液中添加更容易发生氧化还原反应的水溶性电子介体,电子介体从阴极上获取电子变为还原态,再扩散到NAD(P)+附近,将电子传递给NAD(P)+,使之转化为NAD(P)H,产生氧化态的电子介体扩散回阴极继续运载电子,实现NAD(P)H的循环再生。使用游离的电子介体的缺点在于更换电解液时难以回收利用,且可能对酶产生一定的毒性[20-21],将电子介体修饰在阴极上催化NAD(P)H再生是可行的解决方法之一。
NAD(P)H再生反应的电子转移过程复杂、副反应多,会对电-酶偶联还原CO2系统的长期运行效率造成不利影响。在电-酶偶联CO2还原系统中使用人工开发的辅因子类似物能使整体反应的可控性更好,是引入NAD(P)H再生反应的替代策略。常见的辅因子类似物为甲基紫精(methyl viologen,MV)及其衍生物,通过在分子两端修饰不同的基团,能够调节辅因子类似物的还原电极电位,并改变其与不同酶之间的亲和性[22-24]。
1.2 NAD(P)H非依赖型系统
部分CO2还原酶的分子表面含有铁硫簇、核黄素和血红素等“电子接收器”结构,或具有暴露的氧化还原中心。基于这种酶搭建的系统可以摆脱NAD(P)H的限制[25],这类系统的电子转移机制分为两类:当酶与电极之间的距离很近时,电极上的电子直接通过隧穿效应到达酶的“电子接收器”或氧化还原中心部分,称为直接电子转移(DET);当酶与电极之间的距离较远时,使用人工电子介体在电极与酶的“电子接收器”或氧化还原中心之间穿梭递送电子,称为间接电子转移(MET)。根据Marcus电荷转移理论,为了保证酶的活性结构与电极之间发生电子隧穿,二者距离应小于1.4 nm[26]。因此,在直接电子转移系统中,必须采用固定化手段将酶负载在电极表面,但酶的用量会受到电极面积的限制,同时,在DET体系中,界面电子转移率通常较低。
电子介体(如2,2′-联吡啶和4,4′-联吡啶衍生物)能够介导酶与阴极间的电子转移,构成MET系统。如,Choi等[27]利用甲基紫精在阴极和甲酸脱氢酶之间的穿梭电子,在5 h内生产了6 mmol/L甲酸盐。Sakai等[28]通过在炭黑修饰的电极上吸附来自Methylobacterium extorquensAM1的甲酸脱氢酶(FDH)MeFDH,以合成的1,1′-三亚甲基-2,2′-二溴联吡啶(TQ)作为高效的电子传递介体,成功制备了气体扩散型生物阴极,用于CO2与甲酸的相互转化;同时发现,电极的疏水性在构建气体扩散型阴极系统中起关键作用,因为它会影响吸附的FDH与可溶性TQ之间的相互作用。此外,典型的介体分子还包括有二茂钴、醌类和吩噻嗪类等。
2 电-酶偶联催化转化CO2的研究进展
目前,在酶促电催化系统中用于直接转化CO2的酶包括碳酸酐酶(carbonic anhydrase,CA)、一氧化碳脱氢酶(carbon monoxide dehydrogenase,CODH)、甲酸脱氢酶(formate dehydrogenase,FDH)、氮酶以及巴豆酰辅酶A羧化酶/还原酶(crotonyl-CoA carboxylases/reductase,Ccr)等。这些酶系统还原CO2的反应式见表2。此外,国内外研究者还构建了丰富的多酶级联系统,以进一步利用还原产品,生成更多高附加值化学品。常见的多酶级联系统为FDH、甲醛脱氢酶(formaldehyde dehydrogenase,FaldDH)和醇脱氢酶(alcohol dehydrogenase,ADH)组成的三酶级联系统,可将CO2催化转化为甲醇。也有一些研究者致力于开发酶促电催化CO2还原的多酶级联新途径,将CO2还原的初级产品进一步转化为C6药物前体[29]和氨基酸[30]等。
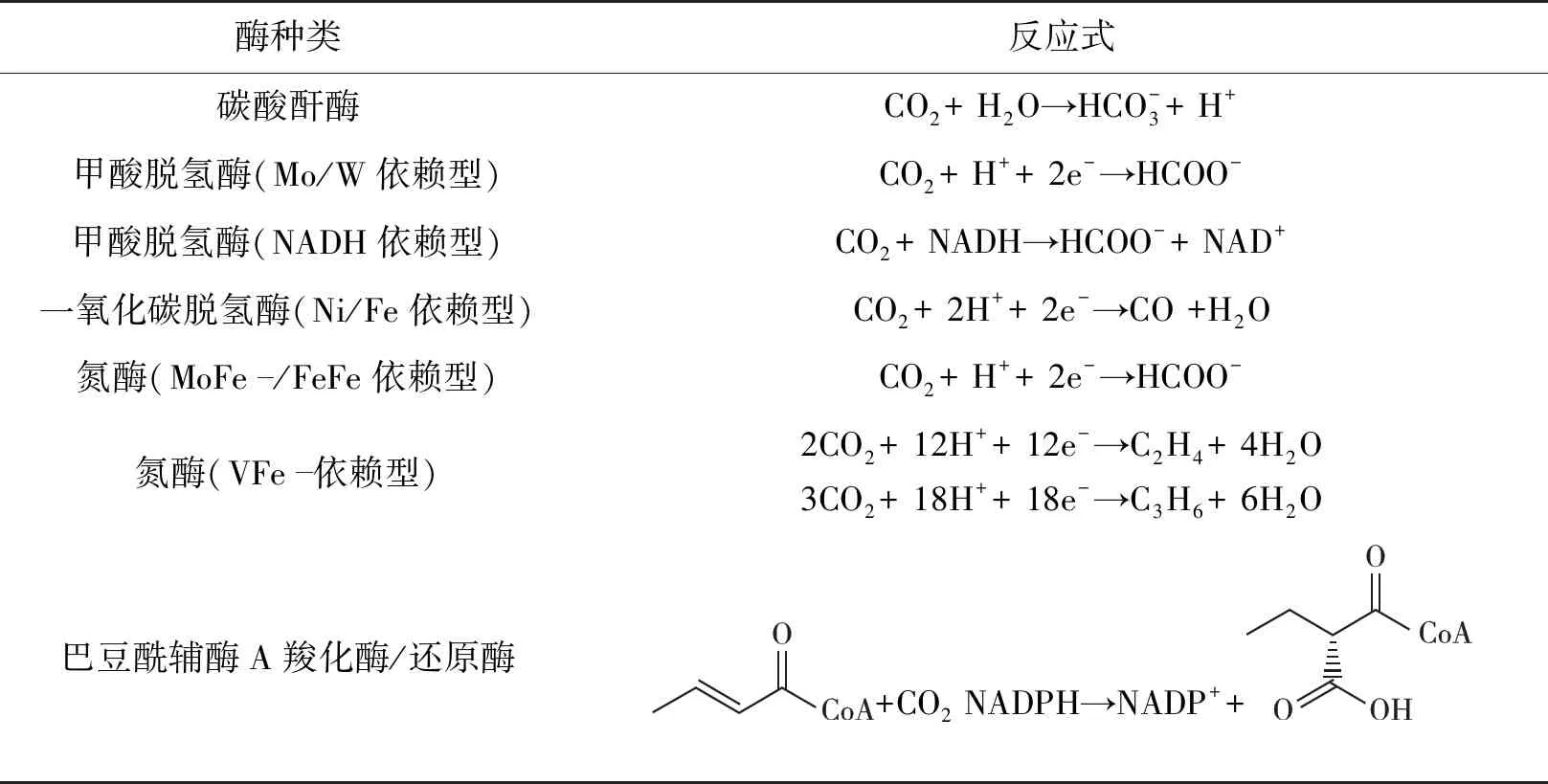
表2 用于直接CO2转化的酶及反应式
2.1 单酶与电极间相互作用的解析
FDH和CODH是最早发现的能够利用电能催化CO2和甲酸、CO2和CO可逆转化的酶。在早期的工作中,研究人员通过向电解液中添加甲基紫精,使之穿梭于电极和酶的氧化还原中心之间传递电子[31-32]。循环伏安法(CV)是研究电极与酶之间电子传输机制的有效手段。2007年,Armstrong团队的Parkin等[33]发现,Carboxydothermus hydrogenform来源的Ni-CODH Ⅰ对CO/CO2之间相互转化的氧化和还原反应都表现出强烈的电催化活性;研究不同pH和温度下的CV曲线后发现,Ni-CODH Ⅰ催化CO2/CO氧化还原循环具有电化学可逆特征。随后Hirst团队先后建立起基于W-/Mo-FDH直接电子转移的电-酶偶联系统[34-35],并提出酶与电极间的电子转移可能是依靠铁硫簇完成的。但在单个循环伏安实验中,由于酶的解吸或变性,电流随着时间的延长而降低。
Sakai等[36]将FDH引入介孔碳电极的孔道中,通过循环伏安测试发现,FDH同时表现出直接和间接电子转移的特征,这是由于靠近酶表面的铁硫簇与电极孔道发生相互作用,直接电子转移通信增强,而间接电子转移过程的氧化还原则由部分解离的黄素单核苷酸(flavin mononucleotide,FMN)介导,两种过程共同促进CO2的还原。
蛋白膜伏安法(protein-film voltammetry,PFV)是由Armstrong等[37]开发的专门研究蛋白质氧化还原电化学的技术,该技术依赖于吸附在电极上的活性蛋白质单层薄膜,且蛋白中应至少有一处氧化还原辅因子与电极紧密结合,以使蛋白质中的活性位点能够与电极进行电子交换[38]。通过向蛋白质薄膜电极施加受控电极电势,可以使用各种伏安技术表征蛋白质的氧化还原特征,获取目标蛋白的多种热力学和动力学信息,从而解析复杂的电子转移机制[39]。Li等[25]通过蛋白质膜伏安法首次明确了来源于Clostridium ljungdahlii的W-FDH中[4Fe-4S]2+/+簇的还原和氧化特征,稳定的蛋白质膜与快速的电子转移使该系统的转化频率(TOF)达到了1 210 s-1。
将PFV与石英晶体微天平(quartz crystal microbalance,QCM)、原子力显微镜(atomic forcemicroscope,AFM)和衰减全反射红外光谱(attenuated total refraction infrared spectroscopy,ATR-IR)等技术相结合,能够更清晰地解析酶和电极之间的相互作用。Lacey团队的Alvarez-Malmagro等[40]通过酰胺键将来源于Desulfovibrio vulgarisHildenborough的甲酸脱氢酶(DvH-FDH)共价固定在金电极和低密度石墨电极表面,并通过修饰电极上的带电基团调节酶的取向(图2),经AFM和QCM分析发现:DvH-FDHs在电极表面形成了紧密锚定的蛋白层;在电解液中加入苄基紫精后,电化学测试能够同时检测到直接和间接电子转移的电流,其中基于间接电子转移的催化电流明显高于直接电子转移的,因此他们认为,除与电极直接接触的单层酶以外,电极顶部仍有许多交联酶分子,这些酶分子只能通过电子介体实现催化作用。以Lacey团队的研究为基础,Fera等[41]近期设计了4-氨基苯基修饰的多壁碳纳米管电极,并将DvH-FDH通过静电作用吸附在电极表面,形成了以利于电子传输的取向,同时进一步使用聚乙烯亚胺稳定了这种取向,结果发现:多壁碳纳米管的引入提高了电化学活性面积,PEI则通过保护酶和增加局部CO2浓度两种效应提高了催化电流,催化电流密度达-230 μA/cm2,能够连续运行11 h。

图2 使用带电基团调节酶的取向[40]Fig.2 Using charged groups to modify the orientation of enzyme[40]
除了电子转移方式外,酶与电极间的结合作用力也能通过PFV和QCM等方法联合表征。Reisner团队的Miller 等[42]通过蛋白质膜伏安法观察到了固定在介孔掺铟氧化锡(ITO)和介孔TiO2电极上的DvH-FDH中[4Fe-4S]簇高效的电荷转移;进一步用QCM分析了酶与介孔TiO2在不同缓冲液离子强度下的相互作用强度后发现,随着缓冲液离子强度的增加,仍有70%~60%的酶被吸附,证明了FDH与电极之间存在比静电相互作用更强的亲和力;同时,用ATR-IR验证了吸附态FDH骨架结构的稳态。2022年,Reisner团队的Badiani等[43]以一系列带电和中性的自组装单分子层来修饰金电极,赋予电极不同的氢键供体能力,并使用QCM分析蛋白质膜在不同氢键作用力下的损失后发现,酶与电极之间DET活性下降的主要是由酶的解吸损失引起的,而仅靠静电作用不足以防止这种损失;同时发现,如氢键作用力等静电作用以外的非共价相互作用对稳定蛋白质膜有重要影响,该结果为建立酶-电极界面提供了理论支持(图3)。此外,Badiani等[44]还构建了具有正电(—NHMe2+)和负电(—COO-)基团修饰的碳纳米管/碳量子点材料,使得吸附在电极上的酶存在不同的取向,以研究材料表面工程对酶的结合和催化CO2还原的影响,结果发现:当使用正电修饰的碳纳米管时,CO2和甲酸可逆转化的电流远远高于使用带负电的碳纳米管;向溶液中添加甲基紫精后,后者才能检测到催化电流,表明当电极带负电时,酶的铁硫簇定位在电极的远端,二者之间的电子交换只能通过电子介体来完成。
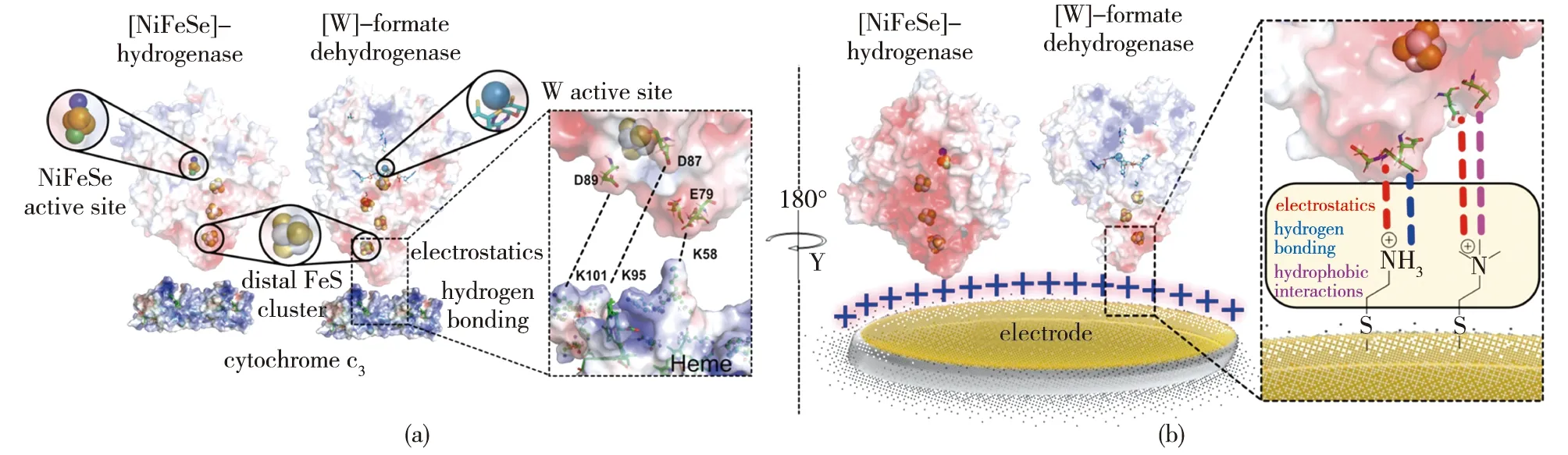
图3 酶与电极间的非共价相互作用[43]Fig.3 Non-covalent interaction between enzyme and electrode[43]
2.2 合理构架电极,提升能量传递效率
NADH依赖型甲酸脱氢酶由于其来源广泛并且高度耐氧而备受关注,然而受限于NADH的应用成本,该类系统难以投入工业规模的长期生产。电化学催化是一种经济、绿色的NADH再生方法,在此过程中,具有催化活性位点的阴极材料在外加电位的驱动下,催化NAD+的烟酰胺部分加2e-和1H+,生成还原型的NADH。为了实现电催化NADH再生,常用的策略是在电极基底上修饰聚中性红[45-48]、Rh复合物电子介体[49-51],或向溶液中添加甲基紫精[52]、中性红[53-54]等游离的电子介体。此外,Cu基材料也作为NADH再生的电催化剂应用在电-酶偶联CO2还原体系中。2018年,Barin等[55]使用泡沫铜电极还原NAD+,能够在240 min内将大部分NAD+还原为NADH。2019年,Song等[56]将Cu纳米颗粒沉积在碳毡上,结果发现:该电极催化NADH再生的性能高于泡沫Cu;为了提高NADH在电极与酶之间的穿梭效率,利用Cu纳米颗粒与酶表面半胱氨酸残基的配位作用将酶固定在电极上,同时连接聚乙二醇摆臂与NADH,以限制NADH的扩散范围(图4(a))。

图4 铜纳米颗粒催化和FNR介导的NADH再生系统Fig.4 Cu nanoparticle catalyzed and FNR-mediated NADH regeneration system
铁氧还原蛋白-NADP+还原酶(ferredoxin NADP+reductase,FNR)在天然光合作用中承担着光反应电子转移的最后一步——将NADP+还原为NADPH[57],通过将FNR固定在电极上,能够介导NADPH的再生。Morello等[58]设计了具有纳米孔道的ITO电极,在该电极上同时限域固定了FNR以及NADP-苹果酸酶(NADP-ME),其中FNR负责接收电子、催化NADPH再生,而ME则承担着CO2同化的功能,最终将CO2和丙酮酸转化为苹果酸(图4(b))。2021年,Castaeda-Losada等[59]在氧化还原水凝胶内共固定FNR与巴豆酰辅酶A羧化酶/还原酶,实现了NADPH的循环再生和巴豆酰辅酶A分子β位的立体选择性羧化,最终的法拉第效率高达92%±6%。
NAD(P)H非依赖型的酶通过铁硫簇等辅因子基团与电极交换电子,当酶被固定化时,酶的氧化还原中心或电活性辅因子必须朝向电极才能发生有效电子传递[60],为消除蛋白与电极间的非电活性接触,研究人员开发了用于固定化酶的氧化还原活性聚合物。这种聚合物将酶包封在电极上,在蛋白四周形成紧密的三维电子传导层,将电极上提供的电子运送到酶的活性位点。
Milton团队的Yuan等[20]设计了一种含有二茂钴(cobaltocene,Cc)结构的低电位氧化还原聚合物用于固定Escherichia coli来源的Mo-FDH,结果发现:通过对聚合物施加-0.66 VvsSHE的电压,Mo-FDH无须利用电子介体就能保持连续的CO2还原活性,在经过500次连续CV扫描之后,还原峰电流下降幅度不到5%。Kuk等[61]则采用了无金属离子的导电聚苯胺水凝胶固定Clostridium ljungdahlii来源的 W-FDH,结果发现:聚苯胺水凝胶电极连续分层的大表面积纳米纤维网络赋予该电极较短的电子扩散长度和纳米尺度的开放通道,在提升酶载量的同时保证了电子递送和底物产物扩散;体系将CO2转化为甲酸的过电位低至40 mV,法拉第效率为92.7%。
由于CO2还原过程通常伴随着质子的快速消耗,电极局部pH与主体相pH往往会产生巨大差异。因此,在设计电-酶偶联CO2还原系统时,应当考虑电极局部微环境的快速变化对酶活的影响。设计合适的缓冲策略,可使酶在电解池中催化作用更高效。最近,Reisner团队的Edwardes Moore等[62]基于生物电催化的有限元模拟(finite element model,FEM)对多孔电极内的局部微环境进行了深度研究,结果发现:通过调整电解液、pH和pKa,可获得酶催化的最佳孔内局部pH;大孔电极具有利于H+传质的优点,将电-酶偶联生成甲酸的催化活性提高了18倍。遵循这一思路,该团队的Cobb等[63]构建FDH@介孔电极体系,通过研究碳酸酐酶催化密闭环境中CO2的水合动力学,辅以有限元分析和电化学测定确定了共固定化碳酸酐酶在该体系中发挥的作用:①通过催化CO2和HCO-3的相互转化缓冲局部pH;②调节CO2和HCO-3的浓度。近期,Cobb等[64]基于上述成果设计了模拟蓝藻羧化体的碳酸酐酶-甲酸脱氢酶共固定化系统,通过使用Good′s缓冲液做电解液来替代碳酸酐酶缓冲局部pH的作用,使所有溶解的碳都能被转化为电-酶催化的底物CO2,而处在最佳局部pH微环境中的甲酸脱氢酶则将CO2完全还原为甲酸。该体系最终实现了大气浓度CO2(12 μmol/L)的直接转化。
最近,Moreno等[65]从工作电压、电解液溶解O2量和系统pH稳定性3个方面进行优化,使电-FDH酶偶联催化转化CO2系统达到最佳工作状态:首先,优化了电解池的整体电压以实现最高的库伦效率;然后,引入O2清除剂Na2S2O3以减少由于O2还原产生的额外电流;最后,搭建了pH反馈回路,通过引入pH泵缓冲反应过程中的pH变化,实现了整体系统性能最大化,在-0.85 VvsAg/AgCl电压条件下,甲酸生产强度率达5 mol/(L·h)。
由于电催化CO2还原反应的底物常以气体形式通入电解池,某些产物(如CO和乙烯等)也以气体形式扩散,所以可使用气体扩散层来增强传质有利于气-液-固三相界面接触,增强催化电流[66-67]。如,Szczesny等[68]设计了一种低电位紫精基氧化还原聚合物,该聚合物将W-FDH固定在电极上,而聚合物/酶层则装载在气体扩散层上,这种设计保证了酶与电极间的电子传递,并且在运行长达45 h后仍保留了80%以上的活性;同时,气体扩散层的引入使气态CO2直接作为酶催化的底物,有效克服了气-液-固界面传质阻碍,电流明显高于直接鼓泡的情况。同样,Becker等[69]针对基于CODH的电-酶偶联系统设计了气体扩散电极,利用二茂钴基低电位氧化还原聚合物固定来源于Carboxydothermus hydrogenoformans的CODH Ⅱ,催化电流密度高达-5.5 mA/cm2,电极活性的半衰期超过20 h(图5(a))。
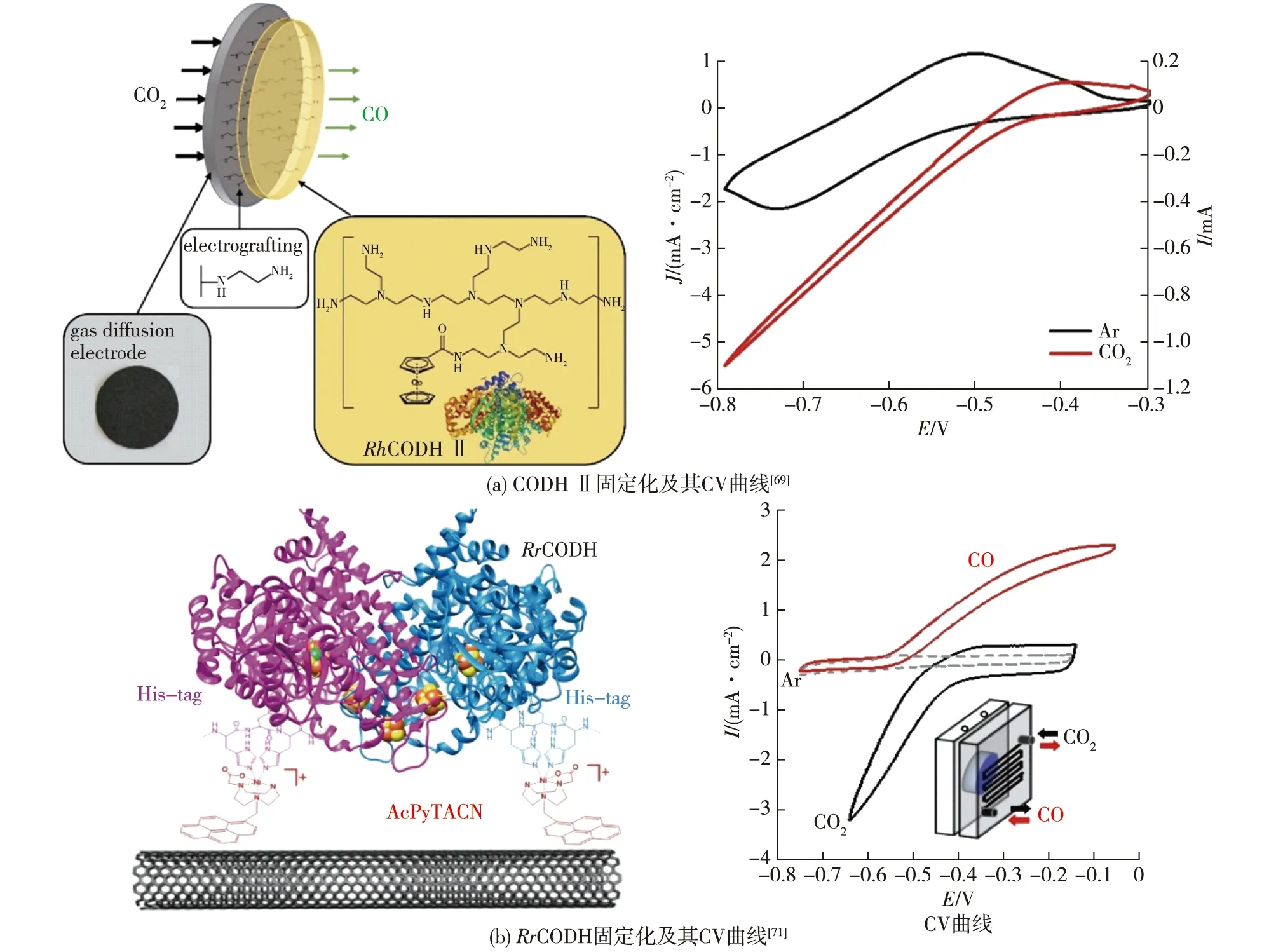
图5 气体扩散电极固定化酶及其对应体系的CV曲线Fig.5 Schematic diagram of immobilized enzyme with gas diffusion electrode and its CV curve of corresponding system
使用一氧化碳脱氢酶时,引入气体扩散电极依然十分有利。Contaldo等[70]将Rhodospirillum rubrum中RrCODH的结构基因cooS、cooC、cooT和cooJ在E.coli中重组表达,并通过聚丙烯酰胺凝胶电泳一步纯化得到高纯度的RrCODH,在实验环境条件下,RrCODH具有显著的活性;然后通过疏水相互作用将RrCODH固定在1-芘丁酸金刚烷酰胺修饰的多壁碳纳米管电极上,在专门设计的CO2/CO气体扩散电解池中,可实现选择性CO2/CO相互转换,转化数(TON)超过80万,并能稳定运行1 h以上。为了进一步稳定酶与电极的结合,Contaldo等[71]合成了一种含有Ni中心的固定分子,使用His-tag将重组RrCODH固定在碳纳米管上,结果发现:在气体扩散生物电极上,生物催化电流达到毫安级,实现了高度稳定的CO2还原(图5(b)),这种设计使RrCODH在催化过程中对氧的耐受性有所提高,在接近零过电位的情况下,TON超过了60万。
2.3 固定化酶技术提高催化稳定性
电-酶偶联系统的另一个挑战在于生物酶蛋白本身的脆弱性,为了保证系统的稳定运行,采用固定化酶技术能够最大程度保持酶的原有结构,减弱局部电场和外界有害物质对酶造成的损伤。
晶态多孔材料(crystalline porous material,CPM)主要包括沸石、金属有机骨架(MOF)和共价有机骨架(COF)等材料[72]。由于晶态多孔材料结构的多样性、均匀可控的孔径和超高的孔隙率,在固定化酶领域拥有巨大的应用潜力[73-74]。
Liu等[53]设计了基于NADH再生的电酶偶联CO2还原体系,NADH再生过程由中性红介导,FDH则固定在聚乙烯亚胺修饰的有序介孔分子筛SBA-15孔道中,结果发现:经固定化的酶催化3 h后的甲酸产量是游离酶的3.7倍;在12个循环后,体系仍保留了80%以上的活性。NU-1006是一种大孔Zr基金属有机骨架材料,其孔径高达6.2 nm,足以容纳甲酸脱氢酶[75]。Farha团队的Chen等[51]使用NU-1006来固定酶,通过结合Rh复合物修饰的电极以循环再生NADH,甲酸的产量与运行稳定性都有了很大的提升。
金属有机骨架UiO-66-NH2具有大比表面积和微孔结构,其孔径更接近CO2的动力学分子直径(0.35 ~0.51 nm),常常被用于CO2的分离和捕集[76-77],所以使用UiO-66-NH2来固定酶,有助于在酶催化之前预先富集CO2底物分子,从而提高底物浓度。然而,UiO-66-NH2的孔径较小,无法直接将酶吸附在孔道内。Jiang团队的Yan等[52]使用合成后配体替代(post-synthetic ligand substitution,PSLS)策略,利用末端配体取代桥接配体向UiO-66-NH2中引入了介孔,并保留原来的部分微孔,形成分级结构,同时保证了MOF将酶固定化和CO2捕集的功能。该系统可利用甲基紫精传递来自阴极的电子为固定化的FDH循环提供NADH,经优化后体系反应3 h的甲酸产量为游离酶的5.57倍,而且72 h后残余酶活保持为初始酶活的70%以上;随后,该团队开发了另一种用于电-酶偶联系统的FDH固定化策略,通过静电作用将FDH预先吸附在聚乙烯亚胺修饰的SiO2纳米花载体上,再进一步包覆ZIF-8形成核壳状结构,这不仅有效防止酶从载体中泄漏,而且ZIF-8的咪唑基团同样对CO2有亲和力,起到预先富集的作用。
Jiang团队的工作为MOF材料在甲酸脱氢酶固定化中的应用提供了有价值的参考,但是MOF材料提升电-酶偶联酶催化效率的机制尚未被解析出来。最近,Yang团队的Jia等[78]使用ZIF-8将来源于Candida boidinii的FDH包封在电极上(图6),结果发现:ZIF-8的存在增加了电极的疏水性和CO2亲和力,最终使甲酸的生产速率比仅使用酶的组别高28倍;原位衰减全反射表面增强红外吸收光谱(attenuated total reflectance surface-enhanced infrared absorption spectroscopy,ATR-SEIRAS)揭示了反应过程中的关键中间体OCHO*(图6(e)),理论模拟表明,ZIF-8中的2-甲基咪唑基团起着类似于辅酶的作用,能够有效调节酶催化位点中缬氨酸的活性,降低基于OCHO*过程的过电位,在-1.1 VvsAg/AgCl的外加电压条件下,复合体系的甲酸产量高达103.9 mmol/(L·h),远远高于仅使用ZIF-8和仅使用FDH的组别(图6(d))。

图6 ZIF-8包封的FDH电-酶偶联系统、表征及甲酸产量[78]Fig.6 Schematic diagram of ZIF-8 encapsulated FDH characterization and formic acid production[78]
2.4 设计电-酶偶联新途径,合成高价值产品
除甲酸脱氢酶、一氧化碳脱氢酶和巴豆酰辅酶A羧化酶/还原酶外,寻找和开发新型生物酶催化剂将为CO2催化转化开拓新方向,进一步提升催化体系的工业应用价值。氮酶的生理功能是将N2还原为NH3,该过程的活化能垒与某些CO2还原反应的能垒相似,故而在特定的非生理条件下,氮酶也能用于催化CO2的还原[79]。2012年,Yang等[80]发现,MoFe-氮酶的双突变体(α-70Val→Ala、α-195His→Gln)具有催化CO2生成甲烷以及还原偶联催化CO2和乙炔生成丙烯的能力。2016年,该团队的Khadka等[81]进一步揭示了氮酶催化CO2还原为甲酸、CO和甲烷的3种作用途径:通过Fe-H的直接氢化物转移,CO2更容易生成甲酸,而当CO2中的C原子直接吸附在Fe位点上时,则更容易生成CO和CH4。2018年,Seefeldt团队的Hu等[82]构建了基于氮酶的电-酶偶联CO2转化系统,使用茂钴基聚合物介导电极向MoFe-或FeFe-氮酶的电子转移,结果发现,二者催化CO2转化为甲酸的法拉第效率分别为9%和32%。Minteer团队的Cai等[83]则发现,在以二茂钴衍生物为电子介体的条件下,来源于Azotobacter vinelandii的VFe-氮酶能够催化C—C键偶联,将CO2转化为乙烯和丙烯,并且催化过程不需要ATP的参与。
Li等[84]通过改变氢键网络和[4Fe-4S]簇的溶剂暴露来调节Hydrogenobacter thermophilus中铁氧还原蛋白的还原电位,并以改造的铁氧还原蛋白作为电子介体向2-氧代酸:铁氧还蛋白氧化还原酶(2-oxoacid:ferredoxin oxidoreductase,OFOR)递送电子,催化乙酰辅酶A和1分子CO2转化为丙酮酸和辅酶A,结果发现:当降低铁氧还蛋白的还原电位降低时,CO2的还原反应在一定程度上能获得更大的驱动力,TOF最大为81.3 min-1,CO2还原活性相当于625 nmol/(min·mg)。
进一步挖掘和改造CO2还原酶,扩展电-酶偶联CO2还原系统的蛋白库是未来的研究方向之一。引入多酶级联系统直接合成高附加值化学品是人们一直以来的热点。用于CO2催化转化最常见的多酶途径为甲酸脱氢酶-甲醛脱氢酶-醇脱氢酶(FDH-FaldDH-ADH)组成的三酶级联系统。CO2在级联反应中经历了由甲酸到甲醛再到甲醇的三步变化。然而,由于三酶级联反应的复杂性以及该体系对NADH的高度依赖,使催化效率不高。为了提升电-酶偶联催化效率,不同的科学家开发了不同手段。如,Addo等[47]发现,加入碳酸酐酶能够加速甲醇生成。Zhang等[50]将3种酶共包封在ZIF-8中,实现了CO2和NADH的预浓缩,并提升了酶催化的稳定性。Zhang等[85]采用另加入天然深共晶溶剂作为电-酶偶联CO2转化的共电解质策略,结果发现,该方法有助于提高CO2溶解度、改善酶的活性。
同时发现,FDH-FaldDH-ADH三酶级联在大表面积电极上可以摆脱对NADH的依赖,通过直接摄取电子的方式催化CO2转化为甲醇。Schlager等[86]利用海藻酸盐-硅酸盐水凝胶将酶固定在碳毡电极上,首次实现了无NADH的三酶级联催化CO2制甲醇,法拉第效率约为10%,然而电子从电极到酶的传递机制尚不清楚。2020年,Seelajaroen等[87]利用羧基化石墨烯共价固定FDH-FaldDH-ADH三酶级联系统,结果发现:在-1.2 VvsAg/AgCl的电压条件下,该系统催化CO2还原时同样不需要NADH的参与,法拉第效率为12%。
开发新型多酶途径,或将酶促反应与其他反应级联提升CO2还原体系配置的灵活性,生产更丰富、更高价值的产品,是构建电-酶偶联催化转化CO2系统的发展趋势之一,包括:①电催化CO2还原-多酶催化偶联;②电-酶催化CO2还原-微生物发酵偶联;③复杂多酶电催化CO2还原系统。
Ren团队的Jack等[29]构建了由CO2到乙醇再到C6药物前体的转化系统:第一步反应由Cu基电催化剂实现催化还原CO2,生成的乙醇经过精馏纯化被输送到多酶反应器中;在酶反应器中,醇脱氢酶首先将乙醇转化为乙醛,随后3分子乙醛被2-脱氧核糖-5-磷酸醛缩酶(2-deoxyribose-5-phosphate aldolase,DERA)利用,合成2,4-二脱氧己糖衍生物;反应24 h后,目标产品高达632~712 mg/L,平均转化率为38%(图7(a))。Chen等[88]验证了电-酶催化CO2还原-微生物发酵偶联的可行性(图7(b)):利用中性红(NR)介导NADH再生,辅助甲酸脱氢酶(FDH)将CO2转化为甲酸,并在电解池中共培养了工程菌Ralstonia eutropha,利用甲酸和CO2生产聚(3-羟基丁酸酯)(PHB);在-0.6 VvsAg/AgCl的条件下,该系统可生成(485±13) mg/L PHB,是对照组(不添加FDH和NR)的3倍。由此可见,中性红在体系中不仅作为催化NADH再生的电子介体,还作为细菌的跨膜电子穿梭载体,将电子直接传递到微生物细胞中,增加胞内还原力。

图7 电-酶偶联催化转化CO2还原系统Fig.7 Electrocatalytic-enzymatic hybrid systems for CO2 reduction
近期,Zhu团队的Wu等[30]利用CO2和NH3为唯一碳源和氮源合成甘氨酸的体外复杂多酶电催化CO2还原系统(图7(c))。该系统依据反应过程中所需的NAD(P)H由Cu基电极再生,并且加入了多磷酸激酶,通过牺牲多磷酸(poly P)再生ATP。整个过程主要包括3个模块的酶促反应:①FDH还原CO2为甲酸;②甲酸依次被甲酸-四氢叶酸连接酶、甲基四氢叶酸环水解酶和亚甲基四氢叶酸脱氢酶催化转化为5,10-亚甲基四氢叶酸;③甘氨酸裂解系统的4种内源酶将5,10-亚甲基四氢叶酸与外源的CO2和NH3缩合生成甘氨酸,四氢叶酸被释放出来。由于电能主要用于NAD(P)H的再生,该系统的法拉第最高可达到96.8%,甘氨酸的生产强度达8.69 mg/(L·h)。
3 结论与展望
电-酶偶联催化CO2还原系统利用可再生的电能为酶促CO2还原反应提供能量,可实现由电能向化学能的转化,从而获得更高的效率和多样化的产品。根据电子转移方式的不同,电-酶偶联CO2还原系统可以分为NAD(P)H非依赖型和NAD(P)H依赖型系统。前者的酶通过直接或间接的方式与电极交换电子,而后者则通过电催化NAD(P)H再生反应为酶提供还原力。本文从电极与酶的互作、电极的理性设计以及新型电-酶偶联系统三个方面对该领域的研究进展进行了系统总结,但该类体系仍处于基础研究阶段,面临着许多挑战。
1)不管是单酶还是多酶系统,在投入电解池之前都需要经过繁琐的分离纯化过程,开发更加简易、系统性的目标酶分离纯化方法对于降低成本、达到规模化生产非常重要。
2)目前应用于电-酶偶联催化CO2还原的酶种类较少,在通过蛋白质工程手段优化已知酶的催化效率、氧耐受性和催化稳定性的同时,还需要探索新的生物酶催化剂,建立和拓展CO2还原酶库。
3)由于CO2的溶解度较低,不利于酶催化反应,因此,需要设计新型的电极和反应器,以实现CO2的原位富集,为CO2的高效转化提供保障。
4)人工电催化系统与酶之间存在相容性差的问题,酶在电解液中容易失活,并且存在动力学不匹配的挑战。因此,应进一步改善电解池配置,采用分池设计等方法,优化两者之间的耦合适配。
总之,随着新型电-酶偶联CO2还原系统的不断优化以及新型多酶途径的开发,有望实现以CO2为原料合成各类化学品,这将在能源、化工、医药和食品等多个领域多个层次上拓展第三代生物制造的应用,为实现“碳达峰”“碳中和”的战略目标提供助力。
仅以此文献给尊敬的欧阳平凯院士!
猜你喜欢
心肺血管病杂志(2019年1期)2019-04-22
天津城建大学学报(2018年6期)2019-01-15
中国有色金属学报(2018年2期)2018-03-26
中南大学学报(自然科学版)(2016年2期)2017-01-19
当代化工研究(2016年9期)2016-03-20
中国资源综合利用(2016年7期)2016-02-03
合成生物学(2015年5期)2015-12-19
中国塑料(2015年8期)2015-10-14
电源技术(2015年5期)2015-08-22
石油化工(2015年11期)2015-08-15
