
腈水解酶反应机制与催化性能调控研究进展
2023-10-10 06:41汤晓玲郑仁朝郑裕国
生物加工过程 2023年5期
汤晓玲,郑仁朝,郑裕国
(浙江工业大学生物工程学院浙江省生物有机合成技术研究重点实验室,浙江 杭州 310014)
腈水解酶属于腈化合物代谢酶系中的一种,能够催化有机腈水解生成羧酸和氨[1]。腈水解酶具有独特的化学、区域和立体选择性,反应条件温和,环境友好,是制备高附加值羧酸的“绿色”选择[2]。腈水解酶在医药、环保、化工、食品和纺织农业等领域获得广泛应用[3-8]。浙江工业大学研究团队于2003年参加了欧阳平凯院士任首席科学家的国家重点基础研究发展计划(973计划)项目“生物催化和生物转化中关键问题的基础研究(2003CB716000)”中课题五“腈水合反应和腈水解反应的方法学及催化机理”的研究,对腈水解酶的筛选、改造、表征和催化机制等进行了系统研究,并将其应用于医药和农药等工业化生产,先后建成(R)-扁桃酸、加巴喷丁、普瑞巴林和2-氯烟酸等医药和农药化学品的规模化生产装置,成为国际上腈水解酶研究开发成功的单位之一[9]。随着高端医药、农药和材料对复杂(手性)羧酸化合物需求的迅速增长,腈水解酶在高端精细化学品合成中展现出日益巨大的应用潜力。所以开展腈水解酶催化机制解析及催化性能调控研究,对提升腈水解酶工业应用属性,拓宽其应用范围具有重要意义。
1 腈水解酶的来源与分类
腈水解酶由Thimann等[10]于1964年首次从大麦叶中分离得到,具有水解吲哚乙腈的活性。此外,该酶还对26种不同的腈化合物具有水解活性。由此这类酶被命名为腈水解酶[11]。Hook等[12]以蓖麻碱为碳源,筛选得到产腈水解酶的微生物(来源于Pseudomonas)。此后,以腈化合物为唯一碳源或氮源,在细菌和真菌中均发现了腈水解酶,包括假单胞菌属、不动杆菌属、红球菌属、产碱杆菌属、诺卡菌属等细菌来源的腈水解酶[13-14];赤霉菌、红球线虫、镰刀菌、黑曲霉等真菌来源的腈水解酶[15-17];水稻、烟草、玉米、拟南芥等植物来源的腈水解酶[18-19]。据不完全统计,已发现的腈水解酶来源近50种[20]。
根据腈水解酶催化的腈化合物结构,可将其分为三类[21]:①能够作用于脂肪腈(丙烯腈和戊二睛)的脂肪族腈水解酶(aliphatic nitrilase);②能够水解芳香族腈(苯甲腈)的芳香族腈水解酶(aromatic nitrilase);③能够水解含酰基官能团腈类化合物(酰基乙腈)的腈水解酶(acyl nitrilase)。但进一步研究发现,有些腈水解酶表现出比较广泛的底物适应性,如Rhodococcussp.NDB1165来源的腈水解酶对芳香族和脂肪族腈类化合物均具有水解活性[22];Syechocystissp.PCC6803 来源腈水解酶既对富马腈、丁腈等脂肪族腈类具有活性,同时又对苯乙腈等芳香族腈以及 2-氰基吡啶和3-氰基吡啶等杂环类腈具有催化活性[23]。
2 腈水解酶的催化机制
在对腈水解酶研究的早期,由于腈水解酶晶体结构少,学者们只能通过对腈水解酶序列的比对分析,结合腈水解酶超家族的晶体结构,推测腈水解酶序列和结构特征关系。近几十年来,Syechocystissp.PCC6803 腈水解酶Nit6803(PDB:3WUY)、Pyrococcus abyssiGE5腈水解酶PaNit(PDB:3IVZ)和拟南芥腈水解酶NIT4(AtNIT4,PDB:6I00)等晶体结构被研究解析:腈水解酶单亚基蛋白是一种αββα三明治结构,一般都含有保守的催化三联体Glu-Lys-Cys[24],在溶液中,腈水解酶亚基围绕特殊的“A”界面形成αββα-αββα二聚体结构[25]。该界面之间的盐桥、疏水作用等作用维持了腈水解酶二聚体的稳定[26-27]。腈水解酶的二聚体结构是低聚化形成具有活性的多聚体的主要单元[28]。除了Fusarium solani、Arthrobactersp.J1和Rhodococcus rhodochrousPA-34等少数腈水解酶以单聚体或者二聚体形式存在之外[24,29],大部分腈水解酶具有催化活性的多聚体单元由4~22个亚基组成(呈月牙形、八字形、环形或者C字形)[24-25,30-31]。腈水解酶还包含C界面、D界面和F界面等一些特殊作用界面(图1),如果其A界面、C界面的氨基酸被替换后可引起腈水解酶活性、立体选择性、热稳定性和底物特异性等性能的变化[3,32-33]。
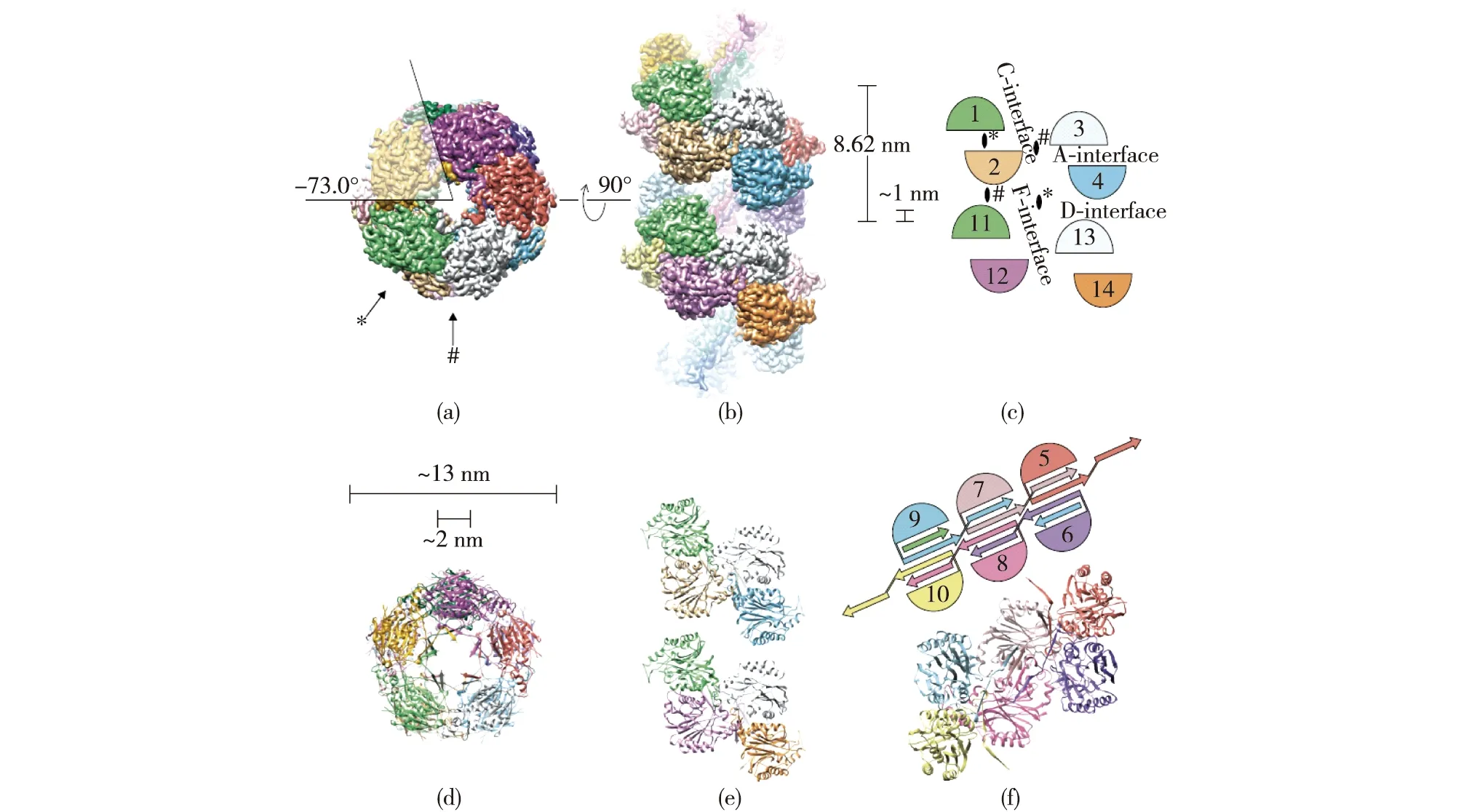
图1 腈水解酶AtNIT4晶体结构以及A、C等特殊界面[34]Fig.1 Crystal structure of nitrilase AtNIT4 and the interfaces(A,C) of the nitrilase monomers in the supramolecular spirals or helices[34]
目前,腈水解酶的催化机制尚未完全明确。最早提出、也最被广泛认可的腈水解酶催化机制认为:腈水解酶一般含有保守的催化位点Glu、Lys和Cys,底物进入活性中心后,Cys 残基上的巯基首先攻击底物氰基上的α碳原子,形成酶-亚胺复合物,随后 Glu 残基提供质子转移给酶-亚胺复合物,氮原子得到质子后消除亚胺结构并释放 NH+4并产生1分子羧酸(图2,pathway A)[35]。在此过程中,Glu增强了对巯基的亲核性,并参与质子转移过程,而Lys则起到稳定四面体结构的作用[36-37]。一般认为,腈水解酶仅会专一性地催化腈水解生成羧酸和氨,但现在研究发现一些腈水解酶同时具有腈水合活性(即催化反应的产物包括酰胺和羧酸)[38-41]。如,Piotrowski等[42]利用拟南芥腈水解酶催化β-氰基丙氨酸,反应产物除天冬氨酸外,天冬酰胺的含量超过60%。此外,在细菌和真菌来源的腈水解酶催化反应的过程中也发现了酰胺的存在。同时,腈水解酶催化形成酰胺的机制与腈水合酶不同。根据推测的机制所示(图2,pathway B),Glu提供的质子与溶剂中的水形成亲核试剂攻击中间体,将质子转移给Cys残基,破坏与氰基α碳原子的硫醇形式而产生1分子酰胺。此外,空间位阻、底物取代基的电负性及反应条件(如温度和pH)等因素都可能影响反应体系中羧酸与酰胺的比例[41,43]。
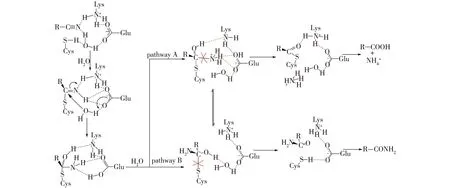
图2 推测的腈水解酶催化机制[44]Fig.2 The proposed mechanism of nitrilase with hydrolysis and hydration activity[44]
Jiang等[40]通过改造Syechocystissp.PCC6803 腈水解酶Nit6803的反应专一性,并对底物-腈水解酶中间四面体复合物的结构分析,对腈水解酶pathway A和pathway B竞争结果提出了新的机制:在腈水解酶催化腈化合物生成中间体后,底物氮原子到催化位点Glu的氧原子的距离和催化位点Glu氧原子到Cys的硫原子的距离会影响羧酸和酰胺的生成比例。Yu 等[5]理性设计Synechocystissp.腈水解酶,使其立体选择性发生反转,并采用这一机制对其突变体酰胺含量的变化进行了分析。
此外,腈水解酶超家族其余分支晶体结构的解析也对腈水解酶催化机制提供了有益的参考。如,通过对Geobacillus pallidusRAPc8的脂肪族酰胺酶(PDB:2PLQ)的晶体结构的研究,Kimani等[45]提出,除了保守的催化位点Glu、Lys和Cys之外,Lys附近保守序列中1个额外的Glu对酰基-酶中间体的水解具有重要作用。同时,这一观点也激发其他研究者的兴趣,如Weber等[46]将来源于Geobacillus pallidusRAPc8腈水解酶的142位Glu替换为Leu或Asp等氨基酸后发现,突变酶对所有底物都失去活性。这是因为上述4个位点皆处于腈水解酶超家族的保守序列,该观点也被一些腈水解酶的相关研究者认可并采纳,如Woodward 等[32]在对腈水解酶底物特异性进行改造后发现,腈水解酶催化位点为E61-K148-E155-C182四联体(图3)。
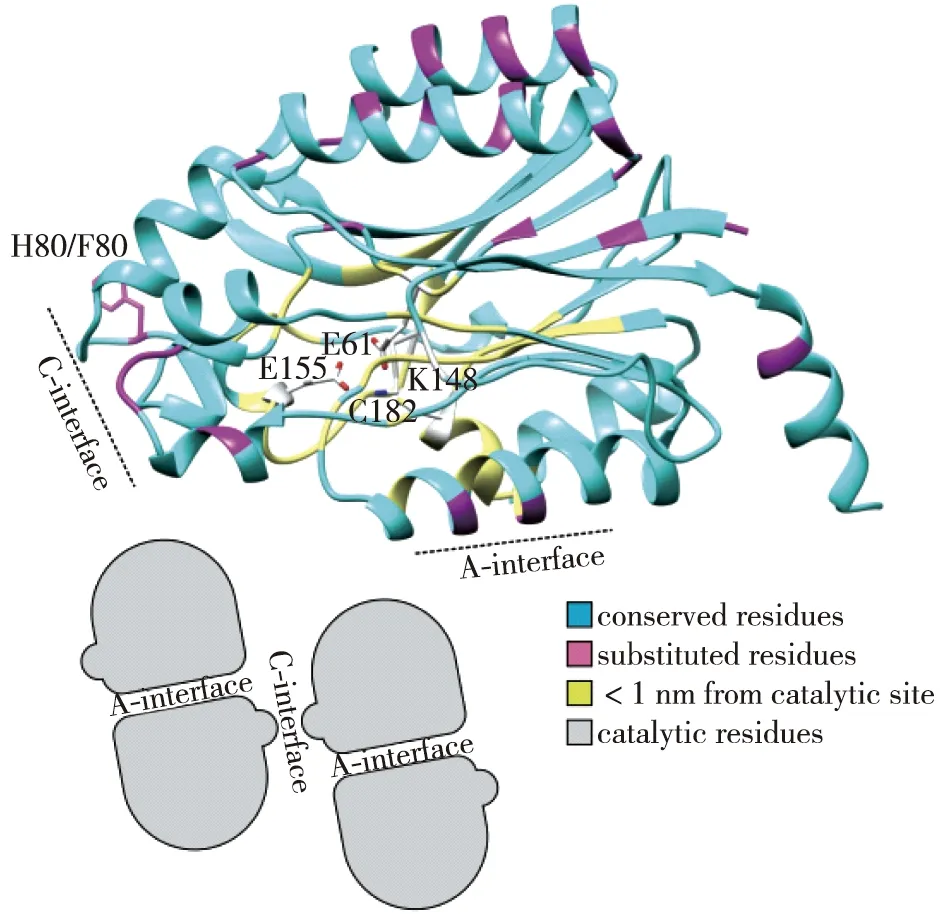
催化四联体为E61、K148、E155和C182图3 植物腈水解酶NIT1结构模型[32]Fig.3 Structural model of a plant NIT1-group nitrilase[32]
3 腈水解酶催化性能调控及应用
由于天然腈水解酶催化非天然底物时活性低、选择性低,且适应工业环境能力弱等原因,限制了其广泛应用,对酶进行调控以提高其应用属性具有重要意义。近二十年来,多个研究团队在系统阐明腈水解酶来源、结构功能关系及作用机制的基础上[11,47],实现了对该类酶多重催化属性的调控。
近年来,国内外研究者们对腈水解酶催化性能的设计改造取得显著进展。浙江工业大学郑院士团队以邻氯扁桃腈为底物,筛选获得了系列具有重要应用潜力的腈水解酶,并通过蛋白质工程提升了来源于Pyrococcus horikoshii腈水解酶的立体选择性,其双突变体T132A/F189T的E值由17.3提升至大于200,且对邻氯扁桃腈的催化活性提升至野生型的4.37倍[4];创制了Acidovorax facilisZJB09122腈水解酶,可催化1-氰基环己基乙腈合成1-氰基环己基乙酸(加巴喷丁中间体),且具有良好的活性、热稳定性和产物耐受性,实现了加巴喷丁的高效酶法合成[48];以来源于Arabis alpina具有较高立体选择性腈水解酶AaNIT和来源于Brassica rapa具有较高活性的腈水解酶BrNIT为亲本,构建了逐段替换式嵌合酶构建技术,获得了活性和立体选择性同步提升的最优嵌合酶BaNIT。在此基础上,通过半理性设计策略确定5个“热点”氨基酸残基并构建饱和突变文库,筛选获得的突变体BaNIT/L223Q/H263D/Q279E的酶活提升5.4倍,E值提高至500以上[49]。此外,国内外其他团队也为腈水解酶催化性能的设计改造,并取得了相当好的结果。如,Wu等[50-51]对A.facilis72W腈水解酶进行改造,结果发现,突变体T201A和双突变体F168V/L201N对羟基乙腈的催化活性比野生酶分别提升了6倍和14倍。Yu等[52]对Syechocystissp.PCC6803 腈水解酶Nit6803进行改造,结果发现,三突变体P149A/I201A/F202V的催化活性是野生型的20.8倍。Gong等[53]利用半理性设计策略提升腈水解酶合成烟酸的催化活性,结果发现,三突变体N40G/F50W/Q207E的催化活性是野生型的2倍。Kobayashi等[54]以吲哚-3-乙腈、噻吩-2-乙腈等为底物时,A.faecalisJM3来源腈水解酶突变体C162N比野生型展现了更高的催化活性。Xie等[55]利用易错PCR技术对腈水解酶AtNIT1进行改造,获得催化活性提高3倍的突变体C236S。DeSantis等[56]对腈水解酶全基因序列进行饱和突变改造,获得了能够在3 mol/L底物浓度下高效催化3-羟基戊二腈的突变体,其产物(R)-4-氰基-3-羟基丁酸是阿托伐他汀的关键手性中间体。
腈水解酶兼具腈水解与腈水合活性,即催化混乱性(catalytic promiscuity)[41-42],使其在生物有机合成中既面临挑战,又蕴含着巨大的潜力。笔者所在的研究团队系统研究了影响腈水解酶反应专一性的关键因素,提出了酶进攻底物特征距离对反应选择性的影响机制,建立了基于特征距离调控实现反应选择性双向调控新方法,获得了催化系列底物严格产生羧酸或酰胺的正、反向调控突变体,并将此调控策略成功应用于多个腈水解酶。野生型Rhodococcus zopfii腈水解酶RZNIT催化2-氯烟腈时,酰胺含量高达88%,基于特征距离调控的改造策略,仅通过一个位点突变(W167G),突变体的水解活性提升至野生型的20倍,催化2-氯烟腈时仅生成2-氯烟酸[37]。同样,Tang等[57]通过锚定Paraburkholderia graminis腈水解酶的关键残基,构建突变文库,筛选获得对2-氯烟腈水合活性消除且水解活性显著提高的优势突变体,成功用于2-氯烟酸的生物合成;利用易错PCR技术对嵌合体腈水解酶BaNIT/L223Q/H263D/Q279E(BaNITM0)进行反应专一性改造,在催化异丁基丁二腈合成普瑞巴林手性中间体的应用时,突变体M127I/C237S副产物酰胺的含量由15.8%降至2.1%,且其催化活性提升至BaNITM0的3.9倍。江南大学的Gong等[38]对真菌来源的腈水解酶进行反应专一性改造,得到的双突变体I128L/N168Q催化反应后,使酰胺的含量由2.8%降低至0.9%,且I128L/N168Q的催化活性是野生型的2倍[58]。此外,Jiang等[40]改造Syechocystissp.PCC6803 腈水解酶Nit6803,结果发现:与野生酶相比,突变体F193N、F193A、F193D和F193Q等的腈水合活性提升20倍以上;以2-氰基吡啶为底物,突变体F193N催化产物中酰胺的比例达73%。Kiziak等[59]对来源于P.fluorescensEBC191的腈水解酶进行改造后发现,当删除其C端后,腈水解酶的酰胺合成能力明显提升,但立体选择性和催化活性显著下降。
在腈水解酶立体选择性调控方面,国内外团队也取得了极大的进展。Lu等[58]聚焦腈水解酶独特的C界面结构,通过定点饱和突变构建突变文库,筛选到的V82L突变体立体过量值明显提升,并与双突变体M127I/C237S进行叠加,可高效、高立体选择性催化异丁基丁二腈;三突变体V82L/M127I/C237S对异丁基丁二腈的E值仍然大于500,催化150 g/L 异丁基丁二腈水解合成普瑞巴林手性中间体(S)-3-氰基-5-甲基己酸,产物的对映体过量值(e.e.值)大于99%。Sun等[3]筛选到对腈水解酶PpL19立体选择性具有重要作用的4个“热点”(M113、R128、A136和 I168),结果发现:野生型对外消旋扁桃腈呈现S型选择性,产物的e.e.值为52.7%;在对上述4个热点进行饱和突变后发现,部分突变体的S型选择性选择性增强,而部分突变体呈现出相反的R型选择性,经过合理组合后,获得的突变体M113L/R128H对S-扁桃腈立体选择性明显增强,产物的e.e.值提升至91.1%;突变体 M113G/A136Y/I168Y则可催化R-扁桃腈,产物的e.e.值为90.9%。Yu 等[5]理性设计Synechocystissp.腈水解酶,使其立体选择性发生反转,并用于光学纯(R)-3-异丁基-4-氰基丁酸的生物合成。Wang等[60]利用易错PCR改造B.cenocepaciaJ2315腈水解酶,结果发现,双突变体I133M/Y199G的立体选择性明显提升,产物光学纯度由89.2%提升至99.1%。Chen等[61]发现,Bradyrhizobium japonicum腈水解酶bll6402NIT的突变体W57F/V134M、W57Y/V134M对异丁基丁二腈具有高立体选择性,产物 (S)-3-氰基-5-甲基乙酸e.e.值大于99%。
除了嗜热菌来源的少数腈水解酶外,大部分腈水解酶都需要面对稳定性一般的困境。Xu等[62]针对腈水解酶典型的多聚体结构,深入探究了多聚体形成过程中两个亚基间“A”界面和多个亚基间“C”界面作用力对酶稳定性的影响机制,结果发现:通过加强界面盐桥、氢键、二硫键以及减小亚基对称螺旋之间的距离可显著提升腈水解酶的稳定性;利用稳固腈水解酶界面的氨基酸残基优化策略,在前期腈水解酶BaNIT突变体基础上,在30 ℃下酶的半衰期提高了10.8倍;获得Acidovorax facilis来源腈水解酶的最佳突变体T201F/Q339K/Q343K,在45 ℃下的半衰期比野生型的提高了14倍。此外,还有一些研究对腈水解酶进行固定化,提升腈水解酶的稳定性和使用效率,以降低生产成本。Ni等[63]通过领苯二酚-壳聚糖包埋的方法对腈水解酶进行固定化,所得固定化细胞可连续进行R-(-)-扁桃酸的生产,反应16个批次后酶活还保留80%。Zhang等[64]用戊二醛直接交联腈水解酶大肠杆菌静息细胞,在100 mmol/L 底物浓度下生产R-(-)-扁桃酸,反应15 批后活性无明显损失。Bauer 等[65]采用聚乙烯醇(PVA)包埋腈水解酶细胞制备丙酸,在细胞和PVA 用量分别为1 g/mL和200 g/L时,酶活回收率高达100%,而且在100 mmol/L 底物浓度下可在填充床上连续反应100 min。
底物特异性的改造也是腈水解酶常见应用需求之一。Zhang 等[66]对Syechocystissp.PCC6803 腈水解酶晶体结构的分析后发现,W146位点对于底物偏好性和底物结合口袋的稳定性有着重要的作用,如果将W146进行氨基酸的替换会改变腈水解酶的底物偏好性。Woodward 等[32]对来源于Capsella rubella的2个高同源性腈水解酶(CrNIT1和CrNIT2)进行改造后发现,腈水解酶C界面上的2个对称螺旋会影响腈水解酶的底物特异性,将82位点由Phe突变成His(或者His突变成Phe)后,CrNIT1和CrNIT2的突变体对3-丁烯腈和6-庚腈的活性发生明显的改变,底物特异性也发生转变。
建立工作量合理、高效且快速的突变体筛选方法对于腈水解酶的改造是至关重要的。目前已有的腈水解酶高通量方法可大致分为pH指示剂法、NH3分析法、羧酸分析法和同位素标记分析法等[67]。腈水解酶催化腈类化合物水解成相应羧酸和NH3,产生的羧酸会使反应环境pH下降。基于此,Banerjee 等[68]建立了一种快速比色法,因为指示剂颜色变化与腈水解酶催化产酸的量成正比。NH3分析法则是对腈水解酶反应生成的NH3浓度进行检测,如NH3与次氯酸盐-苯酚在碱性条件下反应可生成深蓝色靛酚,在612 nm下可定量测定[69],但该方法检测时间较长,且只能检测浓度为20~200 μmol/L的NH3。Banerjee等[70]开发NH3分析法,基本原理是NH3可与巯基乙醇、邻苯二甲醛反应生成异吲哚衍生物,在激发波长和发射波长分别为412和467 nm的情况下,通过荧光强度的大小定量分析NH3的浓度,该方法具有反应时间较短、灵敏度高及检测范围广(2.5~1 000 μmol/L),已被广泛采用(图4)。如,Lu等[58]基于此方法,向高通量筛选反应中加入来源于Pantoeasp.YR343的酰胺酶Pa-Ami,使其可高速检测腈水解酶的总活性和水解活性,从而间接测量腈水解酶的水合活性,筛选反应专一性提升的腈水解酶。羧酸法分析法主要是对腈水解酶反应生成的羧酸进行检测,常用的方法包括将邻羟基苯甲腈及其衍生物作为荧光探针检测腈水解酶活性[71]、根据羟肟酸铁反应检测腈水解酶活性[72]以及紫外分光光度法检测法[73]等。DeSantis等[56]以15N标记的假前手性3-羟基戊二腈为底物筛选对映选择性更好的腈水解酶,因为15N标记的R-3-羟基戊二腈会在R型选择性腈水解酶的作用下生成没有标记的对映体单酸,而在S型选择性腈水解酶会生成带有15N标记的S型对映体单酸,通过质谱可对反应产物进行定性和定量分析。

图4 基于NH3浓度的腈水解酶反应专一性高通量筛选方法[58]Fig.4 High-throughput screening assay for nitrilase with improved reaction specificity[58]
4 结论与展望
欧阳平凯院士领导的“973计划”项目“生物催化和生物转化中关键问题的基础研究(2003CB716000)”为腈水解酶生物催化研究提供了强有力的平台,经过20多年的持续努力,我国腈水解酶生物催化研究取得系列重要进展,形成了基础研究、技术开发、工程创新与工业应用紧密结合的鲜明特色,引起了国内外学术、产业界的广泛关注,引领了该领域国际发展方向。
近年来,蛋白质结构预测等技术快速发展,为腈水解酶的设计和改造提供了有效手段。如何高效、快速获得具有高催化活力、稳定性和选择性等性能的腈水解酶仍是其工业应用亟待解决的关键共性问题。因此,关于腈水解酶催化机制和催化性能调控的研究将仍是未来研究的主要内容。我们要大力弘扬欧阳平凯院士“忠诚精实”的科学家精神,致力创新,服务产业,为我国高水平科技自立自强做出新的贡献。
猜你喜欢
生物信息学(2022年1期)2022-04-01
云南化工(2021年10期)2021-12-21
昆明医科大学学报(2021年2期)2021-03-29
建材发展导向(2021年24期)2021-02-12
四川警察学院学报(2019年6期)2019-12-28
科技创新导报(2018年1期)2018-05-07
石油化工应用(2018年3期)2018-03-24
文化产业(2016年6期)2016-10-19
中国塑料(2014年1期)2014-10-17
建筑材料学报(2014年2期)2014-03-11
