
基于转录组测序的红萍SSR和SNP特征分析
2023-10-10 01:29秦煜郑向丽林钟员
福建农业科技 2023年6期
关键词:转录组
秦煜 郑向丽 林钟员


摘 要:為了解红萍在不同氮浓度胁迫下红萍转录组中SSR、SNP分子标记的详细情况,分析转录组数据中分布的SSR和SNP位点。通过提取红萍根和叶的RNA,构建cDNA文库并利用二代测序平台进行高通量测序,利用MISA对测序数据进行SSR特征分析,同时采用软件STAR进行比对并通过GATK进行SNP识别。结果表明:红萍转录组共有9 009个SSR位点,SSR位点频率为30.12%。其中,以三核苷酸重复和单核苷酸重复为主,分别占总比例的43.66%和26.41%。SNP位点有439 217个,包括转换类型(282 532个)和颠换类型(156 685个);转换类型占比(64.33%)显著大于颠换类型(35.67%)。C/T在所有变异类型中所占比率最高,占总量的17.33%;G/A 位居其次(17.01%)。红萍转录组SSR和SNP位点丰富,可为红萍遗传多样性、遗传结构与分化以及功能基因的开发利用等研究提供依据。关键词:红萍;转录组;SSR;SNP
中图分类号:S 511.41 文献标志码:A 文章编号:0253-2301(2023)06-0044-05
DOI: 10.13651/j.cnki.fjnykj.2023.06.006
Characteristic Analysis of SSR and SNP in Azolla imbircata Basedon Transcriptome Sequencing
QIN Yu1,2, ZHENG Xiang-li3, LIN Zhong-yuan1,4*
(1. College of Geography and Oceanography, Minjiang University, Fuzhou, Fujian 350002, China;
2. College of Future Technology, Fujian Agriculture and Forestry University, Fuzhou, Fujian 350002, China;
3. Institute of Agricultural Ecology, Fujian Academy of Agricultural Sciences, Fuzhou, Fujian 350002, China;
4. Fuzhou Technological Innovation Center of Seawater Planting Industry, Fuzhou, Fujian 350002, China)
Abstract: In order to understand the detailed information of SSR and SNP molecular markers in the transcriptome of Azolla imbircata under different nitrogen concentration stress, the SSR and SNP loci distributed in the transcriptomic data were analyzed. By extracting RNA from the roots and leaves of Azolla imbircata, the cDNA library was constructed and the high-throughput sequencing was performed using the next generation sequencing platform. The SSR characteristics of the sequencing data were analyzed using MISA, and the software STAR was used for the comparison and the SNP identification was performed by GATK. The results showed that: there were a total of 9009 SSR loci in the transctiptome group of Azolla imbircata, and the frequency of SSR loci was 30.12%. Among them, the trinucleotide and mononucleotide repeats were the two most SSR type, which accounted for 43.66% and 26.41% of the total repeats, respectively. There were 439217 SNP loci, including the conversion types (282532) and transversion types (156685). The proportion of conversion types (64.33%) was significantly higher than that of transversion types (35.67%). Among all the variation types, C/T had the highest proportion, accounting for 17.33% of the total SNP loci, followed by G/A with 17.01%. The transcription group of Azolla imbircata had abundant SSR and SNP loci, which could provide a basis for the study of genetic diversity and the genetic structure and differentiation, as well as the development and utilization of functional genes.
Key words: Azolla imbircata; transcriptome group; SSR; SNP
红萍,也称为满江红,是一种小型水生浮游蕨类植物,它的叶腔内共生着固氮的鱼腥藻,具有高效的固氮能力。它起源于晚白垩纪之前,在植物进化上处在独特的位置[1];同时,也是地球上生长最快的植物之一,是潜在的重要碳汇[2]。在最佳生长状态时,红萍的倍殖时间仅为2~5 d[3]。红萍既能无性繁殖,又能有性繁殖,孢子果作为红萍的有性器官,一般在一定的季节交替时期生成[4]。红萍具有重要经济价值和生态效益,比如作为人类的食物、动物的饲料、农田中重要生物肥料,以及可应用于药物生产、沼气生产和水体净化等[5]。红萍特别是在家畜饲料的潜在应用价值方面备受农业界的关注[6-7]。福建省农业科学院建设有国家红萍种质资源圃,保存着来自世界各地红萍种质,共有500多份。但是,很少有对红萍种质遗传结构方面、基因功能开发等的研究。红萍主要为无性繁殖,新老品种数量繁多且易混杂,实际应用中有时对一些品种混杂及亲本无法区分和判断。陈坚等[8]指出解决红萍系统学问题需要分子生物学技术手段。SSR和SNP分子标记对于种质资源鉴定和遗传多样性分析尤为有效。因此,十分有必要挖掘可用于红萍种质分类鉴定的SSR和SNP分子标记。虽然细绿萍基因组已经被解析,但其他红萍品种的基因组仍未见报道[2]。红萍在分子生物学水平的研究报道较少,其组学研究也相对滞后。迄今为止,红萍转录组应用在孢子果诱导[9-10]、共生关系[2, 11]、激素信号[12-13]、维管结构发育[14]、高氮胁迫[15]等挖掘候选基因研究。Coppenolle等[16]应用随机引物扩增多态性DNA (RAPD)分析25个红萍的系统关系;Pereira等[17]开发了RAPD分子标记区分不同红萍品种;Sood等[18]利用RFLP构建可靠的指纹图谱鉴别不同的红萍品种;钟珍梅等[19]利用ISSR分子标记对杂交红萍后代进行鉴定分析。虽有上述红萍的分子标记研究报道,但至今尚未有通过转录组测序分析红萍SSR和SNP分布特征的研究报道。其他蕨类植物转录组挖掘分子标记研究值得借鉴。比如,Mossion等[20]研究报道了蕨类植物扇羽阴地蕨转录组范围的SNP分析其倍性及遗传结构。Yang等[21]研究报道了SSR分子标记可用于研究所有水蕨居群的遗传变异及其地理分布模式。本研究基于前期工作中对红萍在不同浓度氮胁迫的转录组数据,使用MISA(MIcroSAtellite identification tool)软件和GATK2分别鉴定所有转录组中的SSR和SNP位点信息,分析其分布及组成特征,将为今后红萍遗传多样性、遗传结构与分化以及功能基因的开发利用等研究提供依据。
1 材料与方法
1.1 转录组测序数据
材料来自福建省农业科学院国家红萍种质资源圃,供试红萍品种为覆瓦状满江红500。培养温度25℃光照14 h/温度18℃黑暗10 h。材料分别用含0、30和90 mg·L-1的总氮浓度(利用NH4NO3配制)处理5 d后,分别采集叶和根,液氮速冻后超低温冰箱保存。提取红萍RNA,构建测序文库,利用Illumina平台测序。去冗余后的Clean data利用Trinity(https://github.com/trinityrnaseq/trinityrnaseq/wiki)以De novo组装策略进行组装,获得Unigene序列
[22]。
1.2 SSR和SNP分析方法
通过MISA(MIcroSAtellite identification tool)工具对筛选得到的1 kb以上的Unigene搜索SSR位点。按重复类型分类,可分为:单核苷酸重复、二核苷酸重复、三核苷酸重复、四核苷酸重复、五核苷酸重复和六核苷酸重復6种类型。
利用Picard-tools v1.41和samtools v0.1.18整理去除重复序列和融合比对的每个样本bam,并通过GATK2对变异位点的功能、位置和突变类型统计,识别SNP位点[23]。
2 结果与分析
2.1 红萍转录组测序及SSR检索结果
完成了18个红萍样品的转录组测序,获得118.29 Gb Clean data,Q30碱基百分比在94%以上。组装共得到100 043条Unigene,其中长度在1 kb以上的Unigene有29 907条。Unigene的N50为1 634 bp,组装完整性较高。基于Unigene库的基因结构分析,其中SSR分析共获得9 009个SSR标记。
2.2 红萍转录组SSR特征分析
2.2.1 红萍转录组SSR位点数量与密度分布 由表1可知,从红萍转录组中SSR重复类型分析结果看,9 009个SSR中,以三核苷酸重复型SSR数最多,达3 933个,占总比例为43.66%,出现频率为13.15%;其次为单核苷酸重复,占总比例为26.41%,出现频率为7.95%。
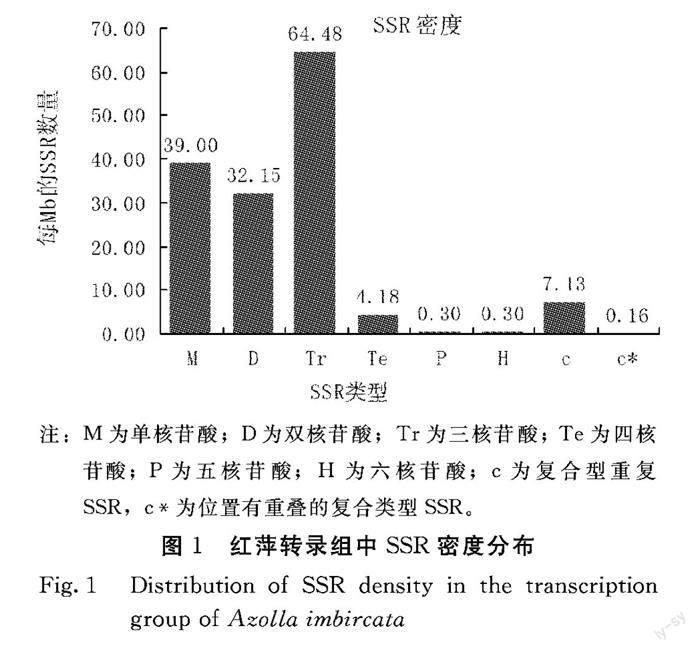
由图1可知,从红萍转录组SSR基序类型分布看,不同类型的SSR密度分布不同。经筛选后的1 kb以上的Unigene序列长度共有61 Mb。本研究发现三核苷酸重复型SSR密度最高,为64.48%,核苷酸重复型SSR密度次之,为39.00%,而五核苷酸和六核苷酸重复型SSR密度最低,均为0.30%。
2.2.2 红萍转录组SSR基序重复类型和长度 由表2可知,红萍转录组中SSR重复数主要分布为5~23次,其中集中在5~15次的SSR占比为99.39%,且重复数随着基序核苷酸数的增加呈现下降趋势,说明SSR重复数存在一定的分布特征。单核苷酸重复类型最多,有12种类型,重复数包括10~20和23,其中10次重复占比最高,占单核苷酸SSR位点的52.96%。核苷酸重复基序有6种重复单元长度,其中含23次重复。
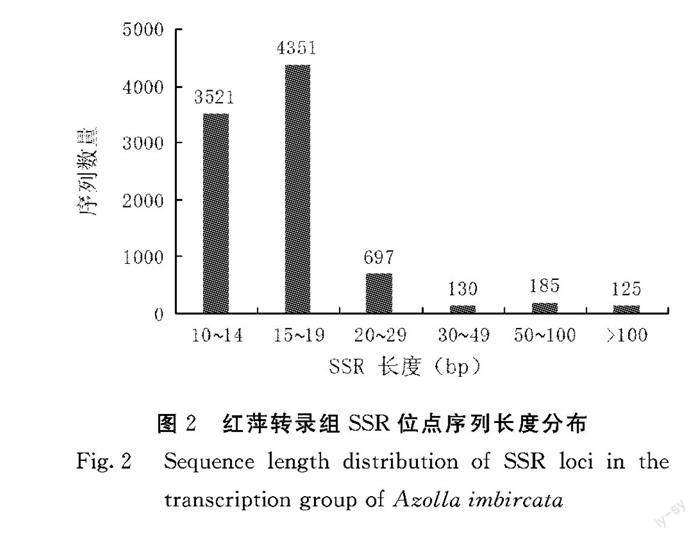
由图2可知,红萍转录组SSR位点序列长度分布在10~222 bp,其中长度在10~14 bp的SSR位点有3 521个,长度在15~19 bp的有4 351个,长度在20~29 bp有697个,长度在30~49 bp的有130个,长度在50~100 bp的有185个,长度>100 bp仅有125个,占比分别为39.08%、48.30%、7.74%、1.44%、2.05%、1.39%(图2)。其中,SSR长度为15 bp的位点数量最多,占总位点的28.64%;长度为18 bp的SSR位点数量次之,占总位点的15.18%。
2.3 红萍转录组SNP位点分析
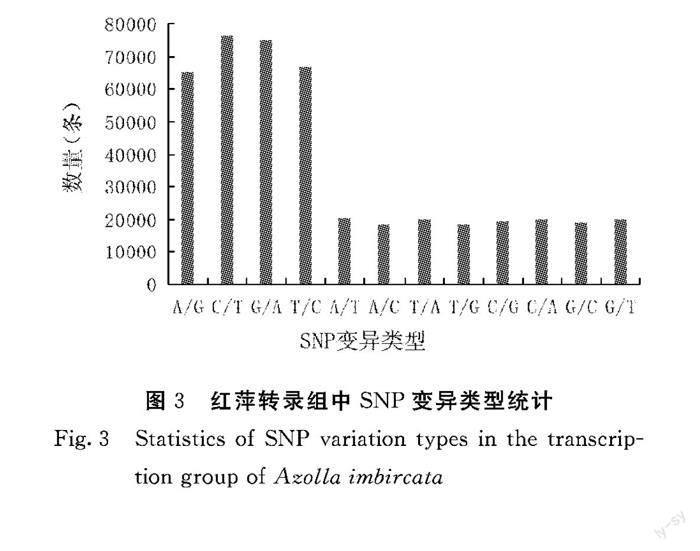
由图3可知,对红萍100 043条序列进行SNP位点识别,成功搜索到SNP位点439 217个,包括282 532个转换类型和156 685个颠换类型;转换类型(64.33%)显著大于颠换类型(35.67%)。C/T在所有变异类型中所占比率最高,占总量的17.33%;G/A 位居其次(17.01%),且这两种变异均属于转换类型。在颠换类型中,A/T所占比例最高,占总量的4.59%;A/C 所占比例最低,占总量的4.20%。
3 讨论与结论
随着测序技术的发展,新一代测序技术为更多物种的基因组研究提供了可能。测序产生的大量数据为挖掘SSR和SNP等分子标记提供了宝贵的资源。本研究利用MISA软件对不同浓度氮胁迫红萍转录组数据进行分析,检测到了9 009个SSR位点。在红萍转录组SSR类型中,三核苷酸重复类型所占的比例最大(43.66%)。三核苷酸重复基序是蕨类植物芒萁的主要SSR基序[24],红萍的该核苷酸重复基序情况与芒萁类似。三核苷酸重复基序含量最高的原因可能是相较于其他的重复类型更加稳定,因为每3个碱基对应翻译成1个氨基酸,生物体为保证蛋白质结构的稳定性,使得序列极少发生移码突变[25]。SSR多态性高低进行分类,SSR序列长度小于12 bp其多态性极低,SSR序列长度在12~19 bp其多态性中等,SSR序列长度大于等于20 bp其多态性较高[26]。SSR多态性的高低是评价其可作为分子标记的标准,而SSR长度是影响SSR多态性的主要因素。红萍的SSR序列长度在15~19 bp的比例为48.30%,推测本研究红萍转录组SSR多态性属于中等,表明后续开发分子标记具有较大的应用潜力。
通过对红萍转录组SNP分析发现,含有439 217个SNP位点,并且转换位点比颠换位点所占比例高。其中C/T转换的比例最高,可能是C易于甲基化,脱氨后转变成T,与前人研究一致。红萍SNP 转换概率约为颠换的1.80倍,小于理论值(转换的概率:颠换的概率=0.5),属于转换偏差现象。类似情况在不少植物中也存在,最早在水稻中发现[27]。出现该现象可能与红萍在水环境中进化过程的选择相关。
综上,利用新一代高通量转录组测序技术,开发红萍SSR和SNP分子標记不失为一种可行的方法。通过大数据筛选通用性高、覆盖度广的分子标记,为红萍种质资源鉴定及功能基因的开发利用提供一定的理论依据。
参考文献:
[1]QIU YL, YU J.Azolla-a model organism for plant genomic studies[J].Genomics, Proteomics & Bioinformatics, 2003, 1(1):15-25.
[2]LI F W, BROUWER P, CARRETERO-PAULET L, et al.Fern genomes elucidate land plant evolution and cyanobacterial symbioses[J].Nature plants, 2018, 4(7):460-472.
[3]ZIMMERMAN W J.Biomass and pigment production in three isolates of Azolla II.Response to light and temperature stress[J].Annals of Botany, 1985, 56(5):701-709.
[4]唐龙飞, 郑德英.我国红萍育种概况及其展望[J].福建农业学报, 1993, 8(1):40-43.
[5]WAGNER G M.Azolla: A review of its biology and utilization[J].Botanical Review, 1997, 63(1):1-26.
[6]KUMAR U, KUMAR A, UMAKANTA N, et al.Nutrient profiling and identification of genetic marker for Azolla sp[C]∥XXIII International Grassland Congress-IGC, 2015; 2015.
[7]KHARE A, CHATTERJEE A, MONDAL M, et al.Effect of supplementing Azolla microphylla on feed intake and blood parameters in growing crossbred female calves[J].Indian Journal of Animal Nutrition, 2016, 33(2):224-227.
[8]陈坚,徐国忠.满江红属系统学研究的新进展[J].植物学通报, 2001,18(4):485-489.
[9]BROUWER P, BRUTIGAM A, KLAHOGLU C, et al.Azolla domestication towards a biobased economy?[J].New Phytologist, 2014, 202(3):1069-1082.
[10]DIJKHUIZEN L W, TABATABAEI B E S, BROUWER P, et al.Far-Red Light-Induced Azolla filiculoides Symbiosis Sexual Reproduction: Responsive Transcripts of Symbiont Nostoc azollae Encode Transporters Whilst Those of the Fern Relate to the Angiosperm Floral Transition[J].Frontiers in Plant Science, 2021, 12:693039.
[11]EILY A N, PRYER KM, LI F-W.A first glimpse at genes important to the Azolla-Nostoc symbiosis[J].Symbiosis, 2019, 78(2):149-162.
[12]DE VRIES J, FISCHER A M, ROETTGER M, et al.Cytokinin-induced promotion of root meristem size in the fern Azolla supports a shoot-like origin of euphyllophyte roots[J].New Phytologist, 2016, 209(2):705-720.
[13]DE VRIES S, DE VRIES J, TESCHKE H, et al.Jasmonic and salicylic acid response in the fern Azolla filiculoides and its cyanobiont[J].Plant, Cell & Environment, 2018, 41(11):2530-2548.
[14]BROUWER P, BRUTIGAM A, BUIJS V A, et al.Metabolic Adaptation, a Specialized Leaf Organ Structure and Vascular Responses to Diurnal N2 Fixation by Nostoc azollae Sustain the Astonishing Productivity of Azolla Ferns without Nitrogen Fertilizer[J].Frontiers in Plant Science, 2017, 8:442.
[15]ZHENG X, LIN Z, LU J, et al.De novo transcriptome analysis reveals the molecular regulatory mechanism underlying the response to excess nitrogen in Azolla spp.[J].Aquatic Toxicology, 2022, 248:106202.
[16]COPPENOLLE BV, WATANABE I, HOVE CV, et al.Genetic diversity and phylogeny analysis of Azolla based on DNA amplification by arbitrary primers[J].Genome, 1993, 36(4):686-693.
[17]PEREIRA A L, MARTINS M, OLIVEIRA M M, et al.Morphological and genetic diversity of the family Azollaceae inferred from vegetative characters and RAPD markers[J].Plant Systematics and Evolution, 2011, 297:213-226.
[18]SOOD A, PRASANNA R, SINGH PK.Fingerprinting of freshly separated and cultured cyanobionts from different Azolla species using morphological and molecular markers[J].Aquatic Botany, 2008, 88(2):142-147.
[19]鐘珍梅,黄勤楼,黄毅斌,等. 7种红萍的生物学特性研究与ISSR分子标记[J]. 热带作物学报, 2011, 32(10):1873-1877.
[20]MOSSION V, DAUPHIN B, GRANT J, et al.Transcriptome-wide SNPs for Botrychium lunaria ferns enable fine-grained analysis of ploidy and population structure[J].Molecular Ecology Resources, 2022, 22(1):254-271.
[21]YANG X, LONG Z, GICHIRA A, et al.Development of microsatellite markers in the tetraploid fern Ceratopteris thalictroides (Parkeriaceae) using RAD tag sequencing[J].Genetics and Molecular Research, 2016, 15(15):gmr7550.
[22]GRABHERR M G, HAAS B J, YASSOUR M, et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J].Nature Biotechnology, 2011, 29(7):644-652.
[23]MCKENNA A, HANNA M, BANKS E, et al.The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data[J].Genome Research, 2010, 20(9):1297-1303.
[24]刘丽婷,温强,黄小春,等.蕨类植物芒萁幼孢子体转录组高通量测序及特征分析[J].林业科学研究, 2016, 29(4):500-507.
[25]METZGAR D, BYTOF J, WILLS C.Selection against frameshift mutations limits microsatellite expansion in coding DNA[J].Genome Research, 2000, 10(1):72-80.
[26]TEMNYKH S, DECLERCK G, LUKASHOVA A, et al.Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential[J].Genome Research, 2001, 11(8):1441-1452.
[27]MORTON B R.Neighboring base composition and transversion/transition bias in a comparison of rice and maize chloroplast noncoding regions[J].Proceedings of the National Academy of Sciences, 1995, 92(21):9717-9721.
(责任编輯:柯文辉)
收稿日期:2023-05-20
作者简介:秦煜,男,1999年生,硕士研究生,主要从事水生植物分子生物学研究。
*通信作者:林钟员,男,1988年生,博士,副教授,主要从事海藻、水生植物功能基因组学研究(E-mail:lzy2019@mju.edu.cn)。
基金项目:福建省自然科学基金项目(2020J01862)。
猜你喜欢
江苏农业科学(2017年18期)2017-11-18
江苏农业科学(2017年18期)2017-11-18
科技创新导报(2017年19期)2017-09-13
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年15期)2017-08-30
中国中药杂志(2017年13期)2017-07-31
中国中药杂志(2017年4期)2017-03-28
中国中药杂志(2017年2期)2017-03-25
中国中药杂志(2017年1期)2017-03-06
中国中药杂志(2016年22期)2017-02-13
