
Ab initio study of chemical effect on structural properties of Ti-Al melts
2023-11-02 08:37YunFeng冯运YanFeng冯艳andHaiLongPeng彭海龙
Chinese Physics B 2023年10期
关键词:海龙
Yun Feng(冯运), Yan Feng(冯艳), and Hai-Long Peng(彭海龙)
School of Materials Science and Engineering,Central South University,Changsha 410083,China
Keywords: Ti-Al alloys,chemical effect,atomic structure,ab initio simulations
1.Introduction
TiAl alloys attract much attention in the application of engineering,e.g.,in aerospace and automotive fields,owing to the lightweight and excellent mechanical properties.[1,2]When quenching the metallic melts at a fast-enough rate, crystallization can be avoided and the amorphous state of TiAl alloys is obtained.TiAl metallic glass (MG) has many unique properties that are preponderant to the crystalline counterparts, especially in mechanical performance and corrosion resistance.[3-5]Structural and dynamic properties of MGs can inherit from those of the liquid state, among which the most significant findings are the liquid-like regions and the associated string-like relaxation process.[6,7]From the aspect of inherency,liquid structure can significantly influence that of the metallic glass.
Investigating the structure of TiAl melts is enlightening in unveiling the structural feature for glass-forming ability or undercooling capability.Icosahedral short-range order (ISRO)is a peculiar local packing structure that is of five-fold symmetry but energetically stable.It was proposed first by Frank to explain the undercooling behaviors of liquids.[8]Existence of the ISRO was verified in diffraction experiment on pure Ti melts,[9,10]and liquid mixtures with Ti.[11,12]And soon after,it was found to be prevailing in metallic glass compounded with Zr and/or Cu.[13]They can even aggregate together to form some backbones or mediate-range order in the glass,thus stabilizing the supercooled liquids, and benefiting the glassforming capability.[14,15]
Emergence of ISRO has also been reported in Ti-Al alloys.Upon cooling down, the population of ISRO grows up,and they can connect with each other by volume sharing to form icosahedral medium-range order.[16,17]In TiAl3metallic glass it has been found that the connecting mode of icosahedra, i.e., the face sharing and vertex sharing, can induce the split of the second peak in pair distribution function.[18]Additionally, the cooling rate is another important factor affecting the connections between icosahedral cluster and its defective structure.[19]However, all these researches are within the frame of classical molecular dynamics(MD)simulations.Reliability of the results from the classical MD simulations strongly depends on the accuracy of the empirical potential utilized.[20-23]Anab initostudy on the structure of Ti-Al melts is desired to elucidate the detailed atomic structure including the ISRO.
Additionally, the alloy with Al element usually displays the feature of chemical interaction, due to the strong affiliation of Al with transition metals.[20,24,25]Structure change influenced by this interaction can be summarized as chemical short-range order (CSRO).For instance, the atomic radius of Al can be changed with composition in Au-Al melt[20]and shortened in Zr-Cu-Al MGs.[26]The affiliating propensity can trigger off the formation of chemically biased clusters in Zr-Ni-Al melts.[21]Inevitably, this chemical interaction can impose nontrivial influence on the formation of icosahedral clusters that is a short-range order (SRO) in topology.However,the interplay between the icosahedral packing and chemical interaction in Ti-Al melts has remained unclear in Ti-Al melts hitherto.
2.Simulation details
To address the aforementioned questions, we conductedab initiomolecular dynamics (AIMD) simulations on Ti-Al alloys,with the Al concentration systematically changed,i.e.,in Ti100-xAlxalloys withx= 0, 20, 40, 60, 80, 100.The simulations were performed by using the Viennaab initiosimulation package (VASP),[27]with the electron-ion interaction described by the projected augmented-wave through using the generalized gradient approximation (GGA)[28]for the exchange-correlation function in the Perdew-Burke-Ernzerhof parameterization.[29]Plane-wave basis was used with an energy cutoff of 270 eV.Only theΓpoints were used to sample the Brillouin zone.Newton’s equations of motion were integrated in the velocity form by using the Verlet’s algorithm in NVT canonical ensembles in time steps of 3.0 fs.The temperature was controlled by a Nos´e thermostat.
A cubic simulation box containing 250 atoms was simulated with periodic boundary conditions applied to all the dimensions.The initial configuration was constructed at 2000 K with all atoms randomly placed in the cubic box.After a shorttime relaxation (about 36 ps) the liquid samples were subsequently cooled down to 1500 K.Atom information is collected at this temperature to investigate the structural evolution with concentration.Cell size was adjusted according to the density measured in experiment.[30]At this density,the resulting pressure of the system does not exceed±4 kbar(1 bar=105Pa).For each alloy, the total relaxation time exceeds 60 ps, with the configurations in the first 12 ps discarded and the left ones used for data analysis.
3.Results and discussion
The microscopic structure of amorphous system can be described by the partial pair distribution function (PDF),gαβ(r), which gives the probability of finding an atom of typeβat a distancerfrom the central atom of typeα.The total PDF can be calculated by the partial ones viag(r) =∑αβ cαcβgαβ(r).
Figure 1 shows the calculated total and partial PDFs in Ti-Al melts with various compositions.The shapes and positions of the first peaks are nearly unchanged with the Al concentrationxAlincreasing.The first peak position is usually considered to be the diameter of the atomR(1).R(1)is found to be around 2.80 °A,2.76 °A,2.81 °A,2.84 °A for total,Al-Al,Al-Ti,and Ti-Ti pairs,respectively[see Figs.1(a)-1(d)].The atomic diameters from Al-Al and Ti-Ti pairs are consistent with the values from Pauling radius,[31]whereRAl=1.429 °A andRTi=1.467 °A.This gives the atomic size ratio about 0.97,indicating a small size discrepancy between these two species.


To quantify the chemical effect,we evaluate the chemical environment in the nearest neighbor shells with the partial coordination number,Zαβ, which is the average neighbor number of neighbors of typeβaround the central atom of typeα.It can be calculated by the partial PDF as follows:
whereρis the average number density,cβis the concentration of atomic typeβ,andrcis the first minimum distance in the corresponding partial PDF.The total coordination number for atom typeαisZα= ∑β Zαβ.In the case of random substitution, the neighbors around some atoms are randomly selected according to the concentration of species.This gives the ratio of coordination numbers in random substitution:Zαβ/Zβ=cα, independent of the type of the central atom.
The calculated ratios of the coordination numbers are shown in Fig.2.The dashed lines are the expectation from random substitution.Clear deviation of the concentration is observed around central Al atoms as seen in Fig.2(a).The coordinating Ti atoms around Al atoms are found to be more than those expected from the random substitution,but the surrounding Al atoms are less than expected.This indicates that Al atoms prefer to connect with Ti atoms but avoid connecting with themselves.The excess value to the random substitution reaches a maximum value atxAl=40.The preferred connection for Ti-Al nearest neighbors and the avoidance for Al-Al ones indicate a strong affiliation between Al and Ti atoms.The affiliation tendency difference should be induced by the chemical interaction,analogous to the reported phenomenon of the concentration-biased clusters in the nearest neighbor shells in Zr-Ni-Al melt.[21]In contrast,the coordinating concentration almost coincides with that of the random substitution around the central Ti atoms [see Fig.2(b)].This implies that the biased concentration is mainly caused by the chemical interaction with Al atoms.Significantly high negative values of experimental data are found for the excess molar volume,which may relate to the concentration-biased connection in Al-Ti melts.[30]

Fig.2.Ratio of coordinating numbers around central(a)Al atoms and(b) Ti atoms as a function of xAl, with dashed lines representing the expected value from random substitution.
It is interesting to see how the geometric packing of local structures,e.g.,the packing of icosahedral clusters(ISRO),can be affected by the occurrence of the chemically preferred short-range order.To this end,we utilize the Voronoi tessellation method to characterize the local structure in the melt.The Voronoi index〈n3,n4,n5,n6〉,wherenidenotes the number ofi-edged faces of the polyhedral cluster surrounding an atom,is used to characterize the packing of the nearest neighbor atoms around a central atom.[33]Based on the Voronoi analysis, the local five-fold symmetry (LFFS) is defined as the fraction of the pentagon faces in the polyhedron:d5=n5/∑i ni.[34]The LFFS has been found to be a good indicator for the mechanical property of metallic glasses.[35]Typically,d5is close to 1 for clusters of icosahedral-like order,and it is close to zero for clusters of crystalline-like order(for which triple or quadruple symmetry dominates).
Distribution probability ofd5is Gaussian-like in the liquid [see Fig.3(a)].The population of perfect icosahedral clusters (d5=1) is small, and the highest percentage is only about 1.8% in Ti40Al60melt.This indicates that the most of icosahedral clusters are distorted in liquid.Interestingly, the population of icosahedral-like clusters(d5>0.5)is higher in the mixture than that in pure metal, while the population of crystalline-like clusters (d5<0.5) is smaller in the mixture.This gives a non-monotonic evolution of the population of the icosahedral-like clusters with Al concentration.As the atomic size ratio is small(=0.97),we conclude that the evolution of atomic packing structure can be dominated by the chemical interaction,rather than the entropic effect from the mixing of two different hard spheres.
Although the LFFS clusters can exhibit the evolution tendency of the population with concentration,an overall ordered or disordered extent in liquid is still unclear.For this,we use the structural entropy or Shannon entropy to measure the local structure diversity in the liquid:[36]
Here,Piis the percentage of Voronoi polyhedron of typeiin the liquid.For the ordered system with only one type of local structure,S=0.For the disordered system where the probabilities of all the local structures are the same,Sreaches its maximum value.
Figure 4(a) shows the composition evolution of the calculated structural entropy in Al-Ti system.Interestingly, a non-monotonic evolution is observed: the structural entropy first decreases then increases, with a minimum emerging atxAl=60.Thus, the liquid structure is more ordered in the middle composition.As alluded above,this relatively ordered structure can be due to the chemical interaction emerging in the middle compositions.In experiment and AIMD simulation, it is found that the mixing enthalpy also shows a nonmonotonic evolution; it first decreases, then increases, with a minimum value reported atxAl≈60,[37,38]coinciding well with the composition of the minimum local structural entropy found here.The most negative mixing enthalpy verifies the strongest chemical interaction at this composition, and can lead to the most ordered local structure.
The chemical interaction may also affect the spatial correlation of local structures.To this end,we calculate the spatial correlation indexCijbetween two polyhedral clusters of typeiandj, which is represented by LLFS (d5) and defined asCij=pij/-1, wherepijis the probability of LFFS of typesiandjbeing the nearest neighbors in the simulated alloys,andis the probability of these two clusters in random mixing.[15]Therefore,a positive value ofCijindicates the preferred correlation between clustersiandj, while a negative value represents the anticorrelation (i.e., the atoms of these two types avoid being nearest neighbors).
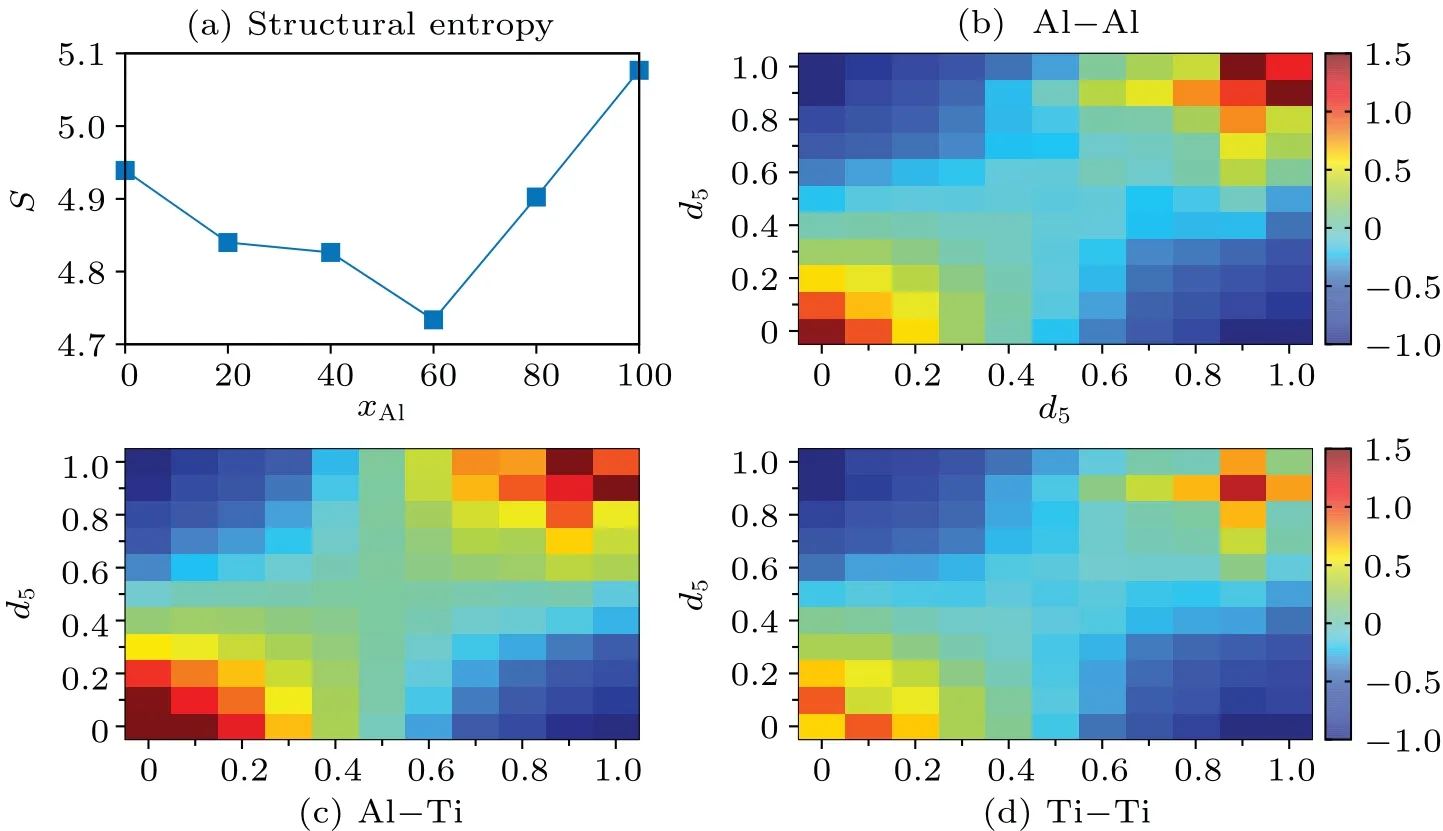
Fig.4.(a)Voronoi structural entropy as a function of composition;patial correlation of different LFFS clusters for the nearest(b)Al-Al pairs,(c)Al-Ti pairs,and(d)Ti-Ti pairs in Ti40Al60.
To distinguish the correlation between different atomic pairs, we calculate the pair-type dependentCi j, with the results shown in Figs.4(b)-4(d) for Al-Al, Al-Ti, and Ti-Ti pairs,respectively.The correlation index is positive in the diagonal direction (from bottom left to top right), which represents the correlation between alike clusters.The largest values appear in the bottom left corner and the top right corner,indicating that the correlation among the icosahedral-like clusters or the crystalline-like clusters is the strongest.Of the different atomic pairs,the Al-Ti pair has the largest maximum correlation extent, Al-Al pair has the intermediate one, and the Ti-Ti pair has the smallest one.This, exactly, corresponds to the chemical interaction found in Fig.2.Although the results shown in Fig.4 are from Ti40Al60melt,we find that this conclusion remains unchanged with the mixing concentration changing.This shows that the chemical interaction plays an important role in determining the connection of local structures in Ti-Al alloys.
4.Conclusions
Ab initiomolecular dynamics simulations are conducted to study the chemical effect on the structural properties in Ti-Al alloys, for which the Al concentration is systematically changed.The liquid structure is measured by pair correlation functions, which shows a splitting tendency in the secondary peaks in the intermediate compositions in Al-Al and Al-Ti pairs.The chemical effect is characterized by the partial coordination numbers, finding that there exists a preferred connection for Al-Ti nearest pairs.This effect is also exhibited as the excess Ti coordinating atoms in the polyhedral clusters:those atoms are characterized by the local five-fold symmetry parameter.With Al concentration increasing,the structural entropy shows a non-monotonic behavior: it first decreases to a minimum value at Ti40Al60,then increases beyond this point.This indicates the relatively ordered local structure induced by chemical interaction.The spatial correlation displays the strongest intensity in the crystalline-like or the icosahedrallike clusters for Al-Ti pairs,coinciding with the preferred connection found in these pairs.
Acknowledgements
This work was done by using the computer resources provided by High Performance Computing Cluster of Central South University.
Project supported by the Open Research Fund of Songshan Lake Materials Laboratory, China (Grant No.2022SLABFN14) and the Natural Science Foundation of Hunan Province,China(Grant No.2021JJ30833).
猜你喜欢
Chinese Physics B(2022年8期)2022-08-31
民间故事选刊(2022年16期)2022-08-23
加油站服务指南(2022年5期)2022-06-02
小学生必读(低年级版)(2021年9期)2021-12-29
政工学刊(2021年12期)2021-12-22
政工学刊(2021年11期)2021-11-01
天工(2021年2期)2021-03-03
小哥白尼(野生动物)(2019年4期)2019-09-10
作文大王·低年级(2018年9期)2018-10-24
作文周刊·小学二年级版(2018年9期)2018-04-18

- Chinese Physics B的其它文章
- Single-qubit quantum classifier based on gradient-free optimization algorithm
- Mode dynamics of Bose-Einstein condensates in a single-well potential
- A quantum algorithm for Toeplitz matrix-vector multiplication
- Non-Gaussian approach: Withstanding loss and noise of multi-scattering underwater channel for continuous-variable quantum teleportation
- Trajectory equation of a lump before and after collision with other waves for generalized Hirota-Satsuma-Ito equation
- Detection of healthy and pathological heartbeat dynamics in ECG signals using multivariate recurrence networks with multiple scale factors