
利用CRISPR/Cas9 技术构建G3BP2 基因缺失型BHK21 细胞系
2023-11-06 08:08陈琳晖屈春惠王世民
现代畜牧兽医 2023年10期
陈琳晖,屈春惠,王世民
( 新疆农业大学动物医学学院,新疆 乌鲁木齐 830052 )
基因编辑是一种针对基因组和转录本进行精确修改的技术,通过核酸酶在基因组的特定位置形成特异性双链断裂,生物体会通过非同源末端连接或同源重组修复断裂双链[1]。成簇规律的间隔短回文重复序列(CRISPR)及相关蛋白酶9(Cas9)构成的CRISPR-Cas9 系统是目前最常用的基因编辑技术,通过特异性的单向导RNA(sgRNA)序列,引导Cas9 蛋白特异结合到靶序列处行使DNA 切割功能[2-3],已广泛应用于各行业并展现出巨大的潜力[4-5]。当细胞受到外界影响,如氧化应激、热休克、病毒感染、紫外线照射时,胞质中会产生致密的颗粒样结构,这种物质被称为应激颗粒(SGs)[6]。SG 主要由RNA 结合蛋白、T 细胞胞质内抗原1(TIA-1)、GTPase 活化蛋白SH3 结构域结合蛋白(G3BP)等成分构成[7]。近年来,一些研究表明,宿主细胞被病毒感染可产生应激反应形成SG,阻止病毒蛋白的翻译,抑制病毒复制[8]。
目前,围绕其中G3BPs的功能研究主要集中于肿瘤以及应激颗粒,G3BP 也作为应激颗粒蛋白被人们所熟知[9-11]。G3BP基因在1996年首次通过与Ras-GAP的SH3结构域的共免疫沉淀被鉴定出来[12],同年从小鼠中分离出第二个与G3BP具有高度同源性的基因[13],这两个基因按照被发现的顺序排列被称为G3BP1 和G3BP2[14-15]。G3BPs主要在RNA 加工和信号传导方面起到作用[16]。作为G3BPs蛋白家族成员,G3BP1 和G3BP2 虽然具有相似的功能,但也各具组织特异性表达。基于此,本研究建立稳定的G3BP2 缺失型BHK21 细胞系,并验证其对细胞抗应激的影响,以期为细胞天然免疫的进一步研究以及研制相关病毒的疫苗奠定基础。
1 材料与方法
1.1 试验材料
1.1.1 细胞株、菌种和质粒本试验所用BHK-21 细胞、DH5α 感受态细胞、pX459载体新疆农业大学微生物与免疫学实验室保存。
1.1.2 试剂、引物与仪器
质粒提取试剂盒(TIANGEN 公司);DNA 提取试剂盒(Omega 公司);Bbs Ⅰ酶、T4 连接酶(赛默飞公司);2×taq plus master mix、lipo 8000 转染试剂、CCK-8、T7 核酸内切酶Ⅰ(上海碧云天生物技术有限公司);GAPDH 一抗、G3BP2一抗、山羊抗兔IgG二抗(Bioss公司)。PCR引物由擎科生物科技有限公司合成。
SCI1000-G PCR仪(美国SCILOGEX公司);垂直超净工作台(苏州净化设备有限公司);恒温培养摇床、C02细胞培养箱(美国Thermo公司);光学显微镜(尼康有限公司)。
1.2 试验方法
1.2.1 sgRNA的设计和寡核苷酸链的合成
根据NCBI 数据库获得G3BP2 基因序列,针对G3BP2基因的两个外显子设计两对sg RNA,送南京擎科生物有限公司合成。利用在线设计网站(http://crispor.tefor.net/)分析得出具体sg RNA得分后,选择得分较高的第1个外显子中的一个sg RNA(sg RNA1)和第2 个外显子中的一个sg RNA(sg RNA2)共两组作为备选序列。所选择的两个外显子均处于所有转录本的共有区域并且位置靠前,可确保试验过程中能够对G3BP2基因的所有转录本进行编辑。在模板的5'端添加CACC,反义链模板的3'端添加AAAC,形成与Bbs Ⅰ酶的黏性末端互补的末端,设计相对应的敲除鉴定引物,引物信息见表1。

表1 引物信息Tab.1 Primer information
1.2.2 敲除载体构建
使用Bbs Ⅰ限制性内切酶切割常用于基因编辑的线性化质粒pX459,并按照凝胶回收试剂盒说明书进行切胶回收。之后将设计的两对sg RNA分别构建到pX459载体上,将退火后的寡核苷酸双链连接上线性化的pX459 载体,转化后挑取单克隆菌落培养提质粒。
退火体系:将合成的引物稀释成浓度为100 μmol/L,各取5 μL的正反链引物溶液。
退火程序:95 ℃ 5 min,室温10 min,冰浴5 min。
连接体系:线性化pX459质粒4 μL、双链sg RNA引物2 μL、10×T4 DNA 连接酶缓冲液1 μL、T4 DNA 连接酶1 μL,补水至10 μL;16 °C过夜。
连接产物转化于DH5α 感受态细胞中,涂布于氨苄抗性固体平板上,37 °C 培养过夜。挑取单菌落扩大培养后作为模板进行菌液PCR,核酸胶验证后,选取阳性克隆送南京擎科生物科技有限公司测序。
1.2.3 敲除载体验证
将BHK-21细胞接种于6孔板,第2 d待细胞长至80%融合度时,将2 条重组质粒G3BP2-sgRNA 分别转染BHK2-1 细胞,转染过程严格按照说明书进行。转染24 h后添加嘌呤霉素,使其终浓度为20 mg/L。在该浓度嘌呤霉素作用下,未转染的细胞会被杀死,之后每天按照该浓度进行换液培养,进行为期7 d 的细胞筛选。取各自存活的部分细胞用于基因组检测,检测引物信息见表1。另一部分细胞使用T7 核酸内切酶Ⅰ酶切验证,选出效果最好的sgRNA重组质粒用于稳定敲除细胞株筛选。
1.2.4 稳定敲除细胞的筛选
使用胰酶消化细胞并进行无限稀释,接种至96孔板培养,将sgRNA1与sgRNA2共转染BHK21细胞,24 h后使用20 mg/L 嘌呤霉素筛选,培养基中嘌呤霉素的浓度可下调至原浓度的一半,细胞不再大量死亡后待细胞长成单克隆细胞群落,取部分细胞用于基因组检测。将有切割修复效果的单克隆细胞长成细胞团后扩大培养,收集部分单克隆细胞和正常细胞,提取细胞总蛋白进行蛋白质印迹技术(WB)检测G3BP2蛋白表达量,选择几乎不表达G3BP2的细胞株继续传代培养。将有切割修复效果及G3BP2 蛋白不表达的单克隆细胞最终转入培养瓶中稳定传代培养。
将2 μmol/L 的二硫苏糖醇(DTT)加入G3BP2-/-细胞株和野生型细胞株中1 h 后镜检观察,以此评估该构建细胞系的抗应激能力。将野生型BHK21 细胞与G3BP2-/-细胞于45 ℃的环境下同样放置1 h,1 h后在显微镜下观察细胞形态,以此测试G3BP2-/-细胞对热应激的反应。
2 结果与分析
2.1 G3BP2-sgRNA载体的构建
针对G3BP2 基因的两个外显子设计并合成两对sgRNA,结果见图1(a);将两对sgRNA 分别构建到pX459载体上,转化后挑取单克隆菌落培养提质粒送至公司测序,测序结果完全正确,结果见图1(b)。重组载体分别命名为:G3BP2-sgRNA1、G3BP2-sgRNA2。
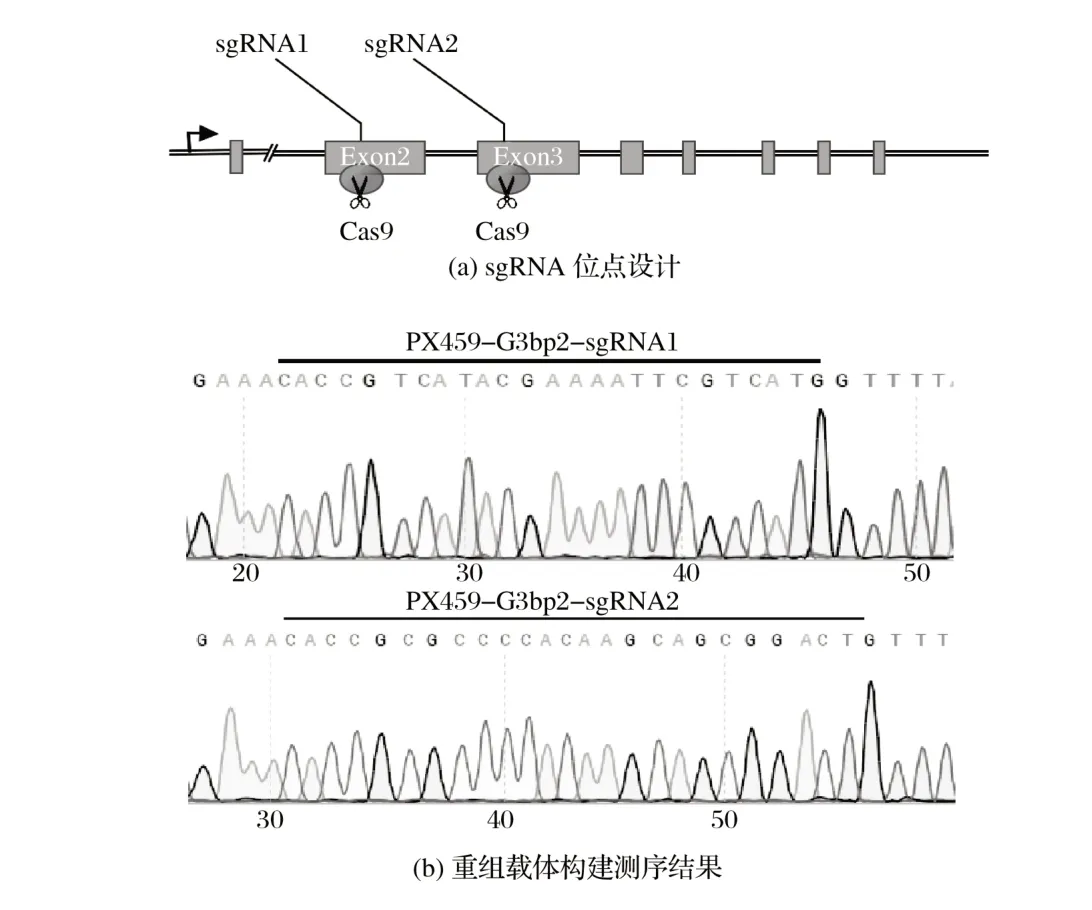
图1 G3BP2-sgRNA载体的构建Fig.1 Construction of G3BP2-sgRNA vector
2.2 G3BP2-sg RNA 质粒转染BHK21 细胞的验证结果(见图2)
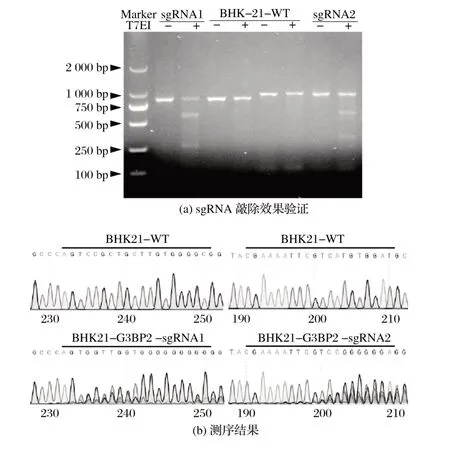
图2 G3BP2-sg RNA质粒转染BHK21细胞验证结果Fig.2 Results of transfection of G3BP2-sg RNA plasmid into BHK21 cells
由图2 可知,将重组质粒分别瞬时转染BHK21 细胞,嘌呤霉素筛选,提取细胞基因组DNA 后使用T7核酸内切酶Ⅰ酶切和测序。序列比对结果显示,sgRNA靶位点处存在杂峰。上述结果表明,重组质粒具有敲除效果。
2.3 稳定敲除G3BP2基因的BHK21细胞系筛选(见图3)
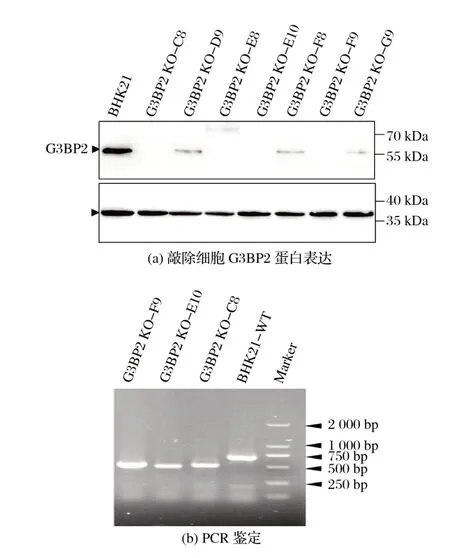
图3 稳定敲除G3BP2基因的BHK21细胞系筛选Fig.3 BHK21 cell line screening with stable knockout of G3BP2 gene
试验共筛选近60株细胞,最后得到7株细胞,将这7株细胞继续传代培养以检测敲除稳定性,在传代培养至十代后提取蛋白进行WB检测,结果见图3(a)。
由图3(a)可知,有3 株细胞(D9、F8、G9)恢复了G3BP2蛋白表达,但蛋白表达量仍显著低于野生型细胞的蛋白表达;有3 株细胞(C8、E10、F9)仍几乎不表达G3BP2蛋白,表明这3 株细胞敲除稳定性良好;而E8 细胞株虽然未表现出蛋白表达,但其在大于70 kD附近存在背景,不排除是非特异性结合。
之后提取C8、E10、F9细胞株的DNA进行PCR扩增及测序,结果显示,敲除细胞株与野生型细胞株相比发生了约220 bp 大小的碱基片段缺失,结果见图3(b)。结果表明,经基因敲除及筛选成功生成了3株G3BP2-/-细胞株。
2.4 敲除G3BP2对细胞应激的影响(见图4)

图4 敲除G3BP2对细胞应激的影响Fig.4 Effect of G3BP2 knockdown on cellular stress
由图4 可知,与野生型BHK21 相比,缺失G3BP2 基因的BHK21细胞对外界环境的变化更敏感。在正常情况下BHK21 的细胞形态为梭形,使用DTT 溶液处理细胞1 h后,缺失G3BP2 基因的BHK21 细胞会缩成近圆形并且细胞膜结构不明显;而野生型的BHK21 细胞仍能够较好地保持梭形的细胞形态,并与正常细胞形态差别不大。即使在PBS 孵育的条件下,缺失G3BP2 基因的BHK21 细胞形态更易受到影响而变成圆形,而野生型BHK21 的细胞形态几乎不发生改变。
之后测试G3BP2-/-细胞对热应激的反应,结果显示,显微镜下观察发现,与野生型细胞相比,G3BP2-/-细胞抵御热应激的能力更弱,表现为细胞发生了聚集、团块化并且大量细胞脱落不再贴壁;而野生型细胞大部分仍可维持贴壁。虽然有研究称,敲低G3BP2不会影响热应激形成的应激颗粒的数量[17],但从眼观细胞表征来看,不论是DTT 造成的应激还是热应激,G3BP2-/-均表现出低抵抗性。
3 讨论
带有荧光标签的EHV-IgD囊膜蛋白表达质粒会使细胞产生荧光斑点。对与gD囊膜蛋白互作的宿主蛋白进一步研究后,本研究猜测产生的荧光斑点为应激颗粒。G3BP1与G3BP2均是参与应激颗粒形成的蛋白,且均可与gD 囊膜蛋白发生相互作用。但有研究表明,敲除G3BP1的小鼠表现出胚胎致死性[18],因而本试验选择对G3BP2进行研究。G3BP2 具有两种亚型,分别是G3BP2a 和G3BP2b[15,19],在WB 检测中有时会出现两个条带,分析原因可能与裂解液的强弱以及裂解液的时间长短有关。本研究使用CRISPR-Cas9 系统对编辑该蛋白的基因进行敲除,针对G3BP2 的两个外显子分别设计了一对sgRNA,使用sgRNA设计网站(http://crispor.tefor.net/),选择评分高且脱靶率较低的sgRNA构建到PX459载体上;该载体可表达Cas9 蛋白,并带有嘌呤霉素抗性,转染后可使用嘌呤霉素筛选阳性细胞。
本研究先使用单gRNA进行敲除,单克隆筛选后进行WB 检测,发现筛选出的细胞株仍能够表达G3BP2 蛋白,测序结果也显示碱基虽然发生了缺失但并未造成移码突变。为提高敲除效果,本研究采取双gRNA共转染产生片段基因敲除的方式,即将两个带有sgRNA的质粒共转染入BHK21 细胞,再重复之前的筛选步骤。DNA 进行核酸胶电泳成像后,结果发现,部分细胞株不止一个条带,其原因可能与二倍体有关。此前,本课题组也曾针对DNA 电泳出现两条带的细胞进行二次单克隆筛选,发现筛选出的细胞仍为两条带。结果表明,sgRNA可能在两条染色体上发生了不同的切割,或与DNA 自身随机修复有关。本试验将几乎不表达G3BP2蛋白的细胞株继续培养数代后,检测这种敲除的稳定性。结果显示,有3株细胞恢复了蛋白表达,但G3BP2 的表达量仍低于野生型BHK21 细胞。有一株细胞在非目的大小附近出现了条带,可能属于抗体非特异性结合,结果成功筛选出几乎不表达G3BP2蛋白的三株细胞,并且敲除稳定性良好。
随后本研究使用DTT对G3BP-/-细胞的抗应激能力进行检测[20],结果显示,在相同浓度、处理时间及细胞密度的条件下,经药物处理后,部分G3BP2-/-细胞形态变圆,部分细胞发生了凋亡,大部分细胞膜结构不再清晰;而野生型细胞的形态表型几乎不发生变化。之后使用热应激测试了细胞,发现热应激同样会对G3BP2-/-细胞造成损伤,使其发生明显的表征变化。上述研究结果表明,G3BP2-/-细胞的抗应激能力明显弱于野生型细胞,这可能与G3BP2蛋白参与细胞抗应激反应有关,缺失G3BP2会导致细胞对无法抵抗多种应激。虽然G3BP2 可能不参与热休克途径的应激颗粒的形成[21-22],但缺失G3BP2的细胞抗应激能力显著减弱,表明G3BP2本身可能并不单纯仅依靠形成应激颗粒保护细胞。
4 结论
本研究利用CRISPR/Cas9 技术成功构建了PX459-G3BP2-sgRNA 重组质粒,并最终得到了稳定的G3BP2-/-细胞系,并通过WB试验在蛋白水平上验证其敲除效率并且验证了其生物学功能。
猜你喜欢
中国现代医药杂志(2020年10期)2020-12-14
食品科学(2018年10期)2018-05-23
现代检验医学杂志(2016年3期)2016-11-15
西南医科大学学报(2015年1期)2015-08-22
医学研究杂志(2015年11期)2015-06-10
医学研究杂志(2015年3期)2015-06-10
特产研究(2015年1期)2015-04-12
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年9期)2015-03-01
中国医药导报(2015年27期)2015-02-28
