
Probing the effect of nitrate anion in CAN: An additional opportunity to reduce the catalyst loading for aerobic oxidations✩
2023-11-18 09:27YingChenChaoChenYonghongLiuLeiYu
Chinese Chemical Letters 2023年10期
Ying Chen, Chao Chen, Yonghong Liu, Lei Yu
School of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou 225002, China
Keywords:Nitrate Alkene Oxidation C=C bond cleavage Ceric Molecular oxygen
ABSTRACT Catalyzed by cerium ammonium nitrate (CAN), the oxidative cracking reaction of alkenes occurred to produce carbonyls in good yields under mild conditions.The reaction employed molecular oxygen (O2) as the safe and clean oxidant.The catalyst dosage was reduced to as low as 0.5 mol%, while no additive was required.Thus, it may afford a generally green synthetic approach for introducing oxygen into organic molecules as well as the biomass degradation and the resource recycling from the C=C bond-containing waste polymers.X-ray photoelectron spectroscopy (XPS) analysis and control experiments demonstrated that the process proceeded via a single electron transfer (SET) reaction-initiated free radical reaction mechanism.In the process, both Ce and NO3- acted as the oxygen carrier to promote the oxidation reaction.The application of the abundantly existed nitrate in CAN was found to be the key for reducing the catalyst loading.
Oxidative cracking reaction of alkenes can allow syntheszing carbonyl compounds, deprotecting key functional groups, and degrading large molecules, especially for those from the biomass[1–11].Thus, it is a significant transformation in many fields such as organic synthesis and biomass utilization, and has attracted comprehensive interests from academia and industry.So far, a series of protocols have been developed.For example, oxidative C=C bond cleavage of alkenes to carbonyl compounds was conventionally achieved using stoichiometric oxidants such as NaIO4,PhI(OAc)2,meta-chloroperoxybenzoic-acid (m-CPBA), or PhIO/HBF4(Scheme 1, method a) [3–6].However, these methods may generate large amounts of wastes, with some being highly hazardous to the environments.Then, reactions using greener oxidants such as TBHP or H2O2were developed [7,8], but these methods still have their own limitations for requiring high loadings of transitionmetal catalysts/ligands, among which some are expensive and toxic.Classical ozonolysis is another frequently employed method(Scheme 1, method b).However, it not only suffers from the unsafe issues, but also suffers from the high cost of the equipments, low ozone generation efficiency associated with the energy waste problem, and the generation of massive wastes during work-up processes [9,10].Moreover, C=C bond cleavage can also be achieved by enzyme-catalyzed oxidative reactions (Scheme 1, method c), but these methods require rather long reaction times and complicated procedures, which lead to low efficiency of the methods [11].
In recent years, catalytic aerobic oxidations have attracted much attention because the abundant molecular oxygen (O2)can be used as cleaner oxidant [12–23].The technique has also been applied in alkene cracking reactions (Scheme 1, method d).Various transition metals were found to be active catalysts, but high pressure of O2, high loadings of the complex and expensive metal catalysts/ligands were usually mandatory conditions[17,18].Methods employing transition metal-free catalysts such as azodiisobutyronitrile (AIBN), 2,2,6,6-tetramethyl-1-piperidinyloxy(TEMPO),N-hydroxyphthalimide (NHPI), CBr4and ArSH were also reported [19–23].Due to the easy decomposition features of most of the organocatalysts, the methods usually require high loadings of the catalysts, which may reduce the practicality and prevent further applications in large scale.Therefore, there is still a great demand in the field to develop practical and efficient methods for aerobic oxidative C=C cleavage that can use a low loading of inexpensive and readily accessible catalysts under mild conditions.
In our cases, we have investigated the oxidative cracking reaction of alkenes by using selenium catalysts, but it requires the use of H2O2oxidant or relatively harsh reaction conditions, or suffer from the incompletely converted substrates [24].Cerium(IV)ammonium nitrate (CAN)-catalyzed aerobic oxidation of alkenes was also reported, but the substrates were majorly styrenes or methylenecyclobutanes, and the use of 5–200 mol% of CAN was required in those works [25,26].Recently, we unexpectedly found that, the nitrate anion (NO3-) in CAN was also a strong catalyst and it could even catalyze the oxidative C=C cracking reaction, so that the CAN catalyst loading could be reduced to be as low as 0.5 mol% (Scheme 1, method e).Herein, we wish to report our findings.

Scheme 1.Methods for oxidative alkene cracking reactions.
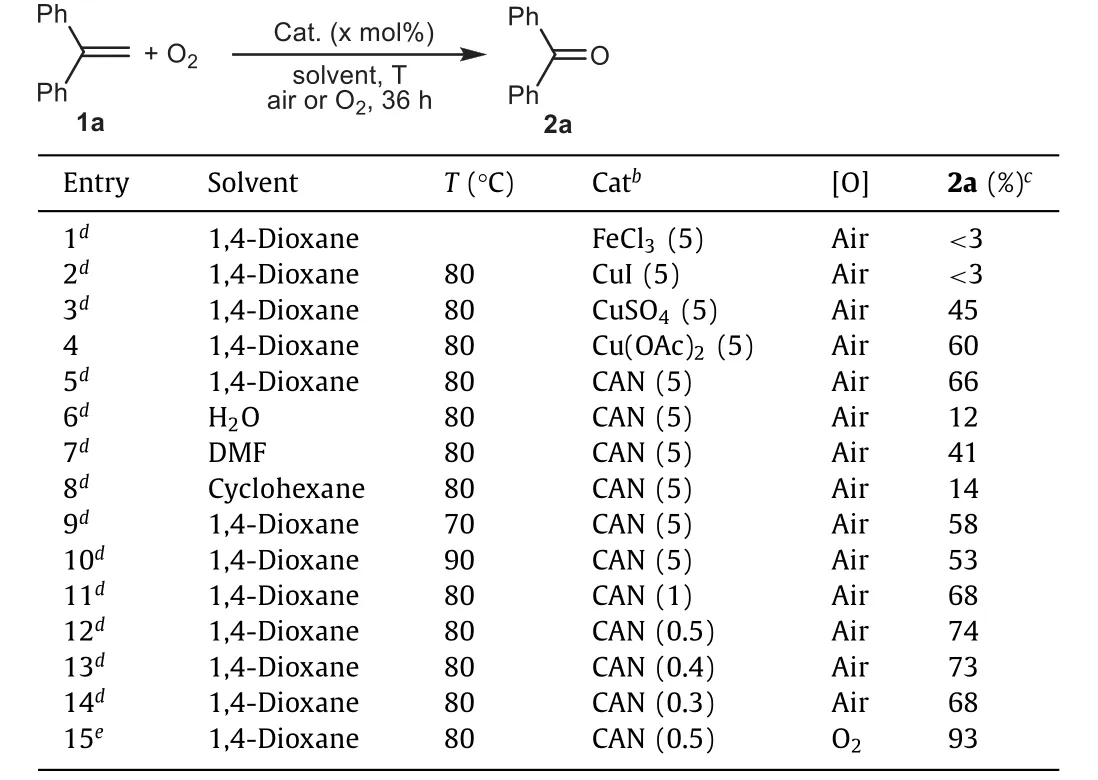
Table 1 Condition optimizationsa.
1,1-Diphenyl ethene (1a) was chosen as the model substrate and FeCl3was initially used as the metal catalyst.However, almost no reaction occurred when heating1aand FeCl3in 1,4-dioxane with air (Table 1, entry 1).CuI was also ineffective for the reaction(Table 1, entry 2), but by using CuSO4or Cu(OAc)2as catalyst, the desired reaction occurred to produce ketone2ain 45%–60% yields(Table 1, entries 3 and 4).CAN was then found to be an even better catalyst, affording2ain enhanced yield (Table 1, entry 5).Besides 1,4-dioxane, other solvents, such as water, DMF and cyclohexane were tested, but all of them led to decreased2ayield (Table 1, entries 6–8).Reducing or enhancing the reaction temperature both led to decreased product yields (Table 1, entries 9, 10vs.5).Using less catalyst did not reduce the product yield and with 0.5 mol%of CAN, the reaction could produce2ain the highest yield (Table 1, entries 12vs.5, 11, 13, 14).In the above reactions, substrate1awas not completely converted and could be observed in thin layer chromatography.Thus, pure O2was then employed as a stronger oxidant instead of air.We were very glad to find that, the reaction was accelerated and could complete within 24 h to produce2ain 93% yield (Table 1, entry 15).
Substrate scope of the reaction was then examined under the optimized conditions in Table 1, entry 15.Results in Table 2showed that the method could be widely applied for the oxidative cracking reactions of a variety of 1,1-disubstituted ethenes.Both electron-enriched and -deficient 1,1-disubstituted ethenes such as1a-1mcould be smoothly oxidized to produce ketones under mild conditions (Table 2, entries 1-13).Introducing an electrondonating group (EDG) into the substrate obviously reduced its activity for the reaction, giving ketone products in decreased yields(Table 2, entries 3, 4vs.1, 2; 7, 8vs.5, 6).In cases of the alkenes1dand1hbearing MeO- as the strong EDG, the reactions were retarded and extended reaction time (48 h) was required (Table 2,entries 4 and 8).Bulky substituents in substrate could also slow down the reaction and only 44%-76% of1i-1kwere converted after 48 h of reactions, but since the unconverted starting material could be recovered, the isolated yields of2i-2kbased on converted substrates were still good (73%-81%, Table 2, entries 9-11).The method was also fit for the exocyclic C=C system in 1-methylene-2,3-dihydro-1H-indene1l, giving 2,3-dihydro-1H-inden-1-one2lin 76% yield (Table 2, entry 12).It also showed some degree of tolerances for the strained rings in substrate and the 16 h reaction of (1-cyclopropylvinyl)benzene1mcould produce the desired cyclopropyl-contained ketone2min 65% yield (Table 2, entry 13).

Table 2 CAN-catalyzed oxidation of 1,1-disubstituted ethenesa.
The reactions of tri- and tetra-substituted ethenes were also tested (Table 3).Introducing a methyl reduced the reactivity of the substrate, and the reaction of1nled to2ain decreased yield(Table 3, entry 1vs.Table 2, entry 1).By enhancing the reaction temperature and extending the reaction time, the reaction of trisubstituted ethene could be improved.For example, heating1nat 100 °C in O2for 48 h led to2ain 80% yield (Table 3, entries 2vs.1).Besides, the ethyl- (1o),n–butyl (1p) or even phenyl substituted substrates (1q) could lead to2ain good yields after a 48 h reaction at 100 °C (Table 3, entries 3-5).The reactions of1r-tproduced2ain 68%-75% yields and the electron-enriched substrate1twas obviously less reactive and was not completely converted (Table 3,entries 8vs.6 and 7).1,1,2,2-Tetraphenylethene1ushowed poor reactivity for the reaction, affording very low substrate conversion ratio (Table 3, entry 9).
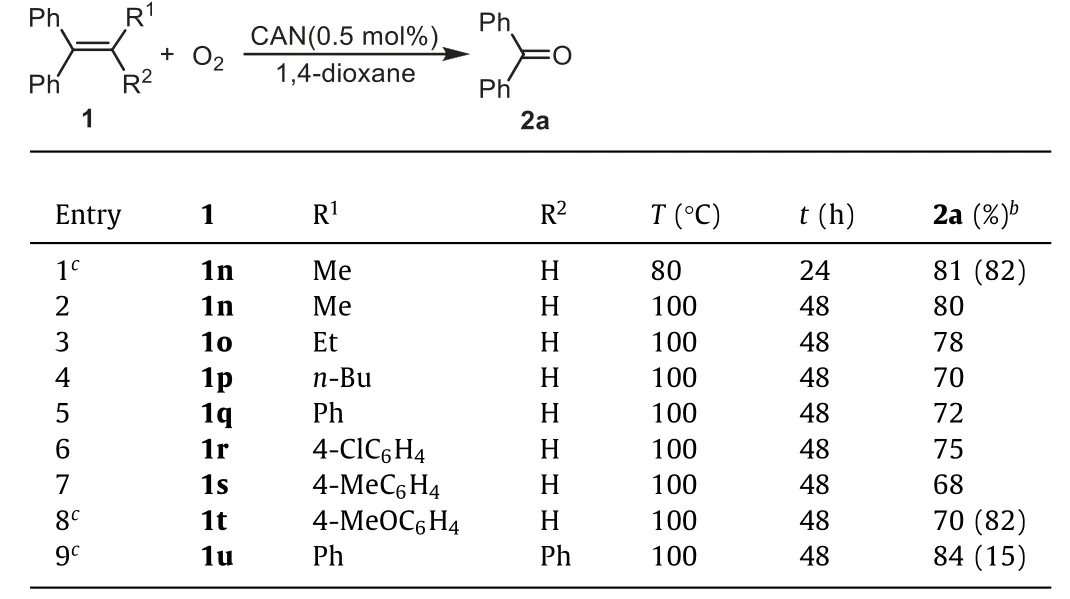
Table 3 CAN-catalyzed oxidation of tri- and tetra-substituted ethenesa.

Table 4 CAN-catalyzed oxidation of styrene and its derivativesa.
The reaction of styrene produced benzaldehyde (2n) in 52%yield, while benzoic acid (3a) was also generated in 40% yield as the unavoidable deep oxidation by-product of2n(Table 4, entry 1).After introducing a methyl on the terminal carbon of styrene, the substrate reactivity reduced and produced2nin 56% yield, while the by-product3ayield decreased (Table 4, entry 2).For the reactions of1xand1ybearing larger alkyl or aryl group, the substrate was not completely converted (Table 4, entries 3 and 4).No reaction occurred when electron deficient substrate1zwas employed(Table 4, entry 5).
Moreover, this CAN-catalyzed oxidative C=C bond cleavage method could be used for the purpose of polyene pollutant degradation (Fig.S1 in Supporting information).The reaction ofβcarotene was chosen as an example to illustrate this idea for its intuitive reaction phenomenon.Heatingβ-carotene in the presence of 0.5 mol% of CAN with O2flow, the color of the reaction liquid faded away gradually (Fig.S1a), while the absorption peak of the samples in UV–vis spectra shifted to the shortwave field (Fig.S1b).These results demonstrated that the C=C bonds inβ-carotene were successfully cut off in this CAN-catalyzed reaction.
Mechanisms were our next concern and a series of control reactions were performed to get the mechanistic insights.The control reaction under N2was performed, but only traces of3awere obtained, while most of the substrate was unconverted, indicating that O2was the crucial oxidant for the reaction (Table 5, entry 1).Using Ce(SO4)2as catalyst resulted in decreased product yield(Table 5, entry 2), which was hardly enhanced even with elevatedCe(SO4)2amount (Table 5, entry 3).Interestingly, after a supplementary of 3 mol% of KNO3that contained the same amount of nitrate as in CAN, the reaction occurred smoothly and produced3ain excellent yield (Table 5, entry 4).The reaction with 0.5 mol%of Ce(NO3)3catalyst produced2ain 90% yield with 95% substrate conversion (Table 5, entry 5).It could be further improved by adding KNO3or using increased amount of Ce(NO3)3(Table 5, entries 6, 7).In the later reaction (Table 5, entry 7), the increased product yield probably attributed to the enhanced NO3-amount other than Ce4+, in accordance with the results in Table 5, entries 2-4.Besides, the nitrate salts of Fe, Cu and Mn, which were all conventional SET catalysts, could catalyze this reaction as well(Table 5, entries 8-13), and the product yields were enhanced after adding KNO3as an additional nitrate source (Table 5, entry 8vs.9; 10vs.11; 12vs.13).KNO3alone could not catalyze the reaction (Table 5, entry 14).By using 3 mol% of HNO3as catalyst, the reaction could produce2ain 75% yield (Table 5, entry 15) and it could also be catalyzed by the KNO3/H2SO4system (Table 5, entry 16).The HNO3-catalyzed reaction could be improved by adding Ce(SO4)2(Table 5, entry 17).The reaction could be restrained by free radical scavenger such as TEMPO or HQ (hydroquinone), indicating that free radicals were involved during the process (Table 5,entries 18) [27–29].In addition, gram-scale reaction was also conducted, and it was found that the yield of this reaction did not decrease significantly after amplification (Table 5, entry 19).

Table 5 Control experimentsa.
X-ray photoelectron spectroscopy (XPS) analysis of the reaction mixtures showed that Ce(IV) in CAN was converted into Ce(III) after reaction (Fig.S2a in Supporting information).Thus, it could be deduced that the reaction was catalysed by transition metals with variable valencesviathe single electron transfer (SET) mechanisms[30,31].Results in Table 5 indicated that nitrate could also promote the activity of the catalytic system, and this could well explain why the catalytic activity of CAN was so high that it could be employed at very low loading (0.5 mol%).In order to explore whether there is hydroxyl radical formation in the reaction process, we designed a verification experiment by taking advantage of the property that salicylic acid can trap hydroxyl radical, and the results obtained were shown in Fig.S2b (Supporting information).After adding the reaction liquid into the salicylic acid sample, a strong adsorption at around 490-650 nm (540 nm at maximum) was observed in the UV–vis spectra (red curvevs.black curve), which was the characteristic peak of the adducts of hydroxyl radical with salicylic acid(i.e.2,3-dihydroxybenzoic acid and 2,5-dihydroxybenzoic acid) reflecting the existence of hydroxyl radical [32].
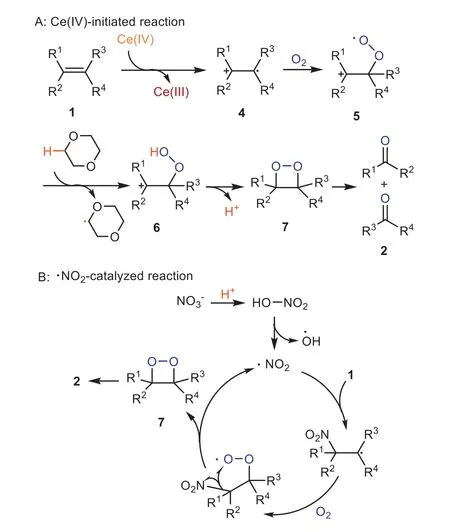
Scheme 2.Possible mechanism of the reaction.
On the basis of the above experimental results as well as the reported literatures [26,33–38], a plausible mechanism was supposed (Scheme 2).The alkenes1were initially oxidized by Ce(IV)to give the active free radical cation4[33].As being confirmed by the XPS spectrum illustrated in Fig.2, Ce(IV) was reduced into Ce(III) after reaction.In the catalysis cycle, the Ce(III) species could be oxidized by O2to regenerate Ce(IV).Oxidation of4with O2led to the intermediate5[34], which grabbed a proton from the 1,4-dioxane solvent and produced the intermediate6[35].Cyclization of6afforded the 1,2-dioxetane7[26] and released a proton.Finally, decomposition of7produced carbonyls as the reaction product.Moreover, the proton generated in the previous step could enhance the acidity of the reaction liquid to facilitate the use of nitrate as an additional catalyst in the HNO3form, which could oxidize alkenes and the generated low valent nitrogen speceis could be reoxidized by O2to produce HNO3and restart the HNO3catalysis cycle B [36–38].The proton releasing during the transition metal catalysis processes was considered to be the key for activating nitrate anion,i.e.convert NO3-into its highly active and oxidative HNO3form.This is why KNO3alone can not catalyze the reaction (Table 5, entrey 14).Therefore, it can be deduced that,CAN is an efficient low-loading (0.5 mol%) catalyst for the oxidative cracking reaction of alkenes because both Ce and nitrate in it are active catalytic species.
In conclusion, we have developed a novel method to split the C=C bond in alkenes by using molecular oxygen as the cheap and clean oxidant.The reaction was catalyzed by low-loading CAN (0.5 mol%) free of any additives.Mechanism studies demonstrated that the ultrahigh catalytic activity of CAN attributed to the fact that both Ce and nitrate may participate the reaction as catalystviathe coupled Ce(IV)-Ce(III) circle and nitroxide circle respectively.The catalyitic activity of anions such as NO3-is a novel finding.It is surprising that the catalytic activity of NO3-is so strong that it can even catalyze the oxidative alkene cracking reaction, which consumes a lot of reaction energy for the high C=C bond dissociation energy (ca.609 kJ/mol).This work not only provides an effi-cient method for carbonyl synthesis, but also leads to a new pathway for biomass degradation, as well as the recycling of the C=C bond-containing waste polymer.It might also inspire new ideas for catalytic system design: the significances of anions in the system should be noticed.Further investigations on the design and application of Ce catalysts are ongoing in our laboratory.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgments
This work was financially supported by Priority Academic Program Development of Jiangsu Higher Education Institutions and Postgraduate Research & Practice Innovation Program of Jiangsu Province (Yangzhou University) (No.KYCX21_3205).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2023.108489.
 Chinese Chemical Letters2023年10期
Chinese Chemical Letters2023年10期
- Chinese Chemical Letters的其它文章
- Tribute text in memoriam of James N.Seiber (1940–2023)
- Recent advances in MXenes-based glucose biosensors
- Oxidative cyclopalladation triggers the hydroalkylation of alkynes✩
- An integrated supramolecular fungicide nanoplatform based on pH-sensitive metal–organic frameworks
- Nickel-catalyzed reductive coupling reaction of monofluoroalkyl triflates with alkyl carboxylic acids toward the synthesis of α-alkyl-α-fluoro-alkylketones✩
- A highly selective fluorescent probe for visualizing dry eye disease-associated viscosity variations