C18环基过渡金属(Os, Ir)单原子对甲烷C—H的活化
2023-11-24 07:11张海平孔雪夏文生张庆红万惠霖
高等学校化学学报 2023年11期
张海平, 孔雪, 夏文生, 张庆红, 万惠霖
C18环基过渡金属(Os, Ir)单原子对甲烷C—H的活化
张海平, 孔雪, 夏文生, 张庆红, 万惠霖
(厦门大学化学化工学院, 固体表面物理化学国家重点实验室, 醇醚酯化工清洁生产国家工程实验室,福建省理论与计算化学重点实验室, 厦门 361005)
由于高度的化学稳定性, 作为天然气主要成分的甲烷分子的转化和利用极富挑战性和研究意义. 然而, 这一问题的突破有赖于甲烷C—H的活化, 特别是温和条件下活化催化剂的研发. 本文采用密度泛函理论方法计算考察了过渡金属TM(Os, Ir)单原子-碳18环(TMC18)对甲烷C—H的活化. 结果表明, 相对于TM单原子, TM单原子键合在碳18环上后, 显著降低了甲烷C—H的活化能垒; 而相较于TM单价阳离子, 则削弱了C—H键裂解物种CH3与TM的结合力. 结合力较弱的TM-CH3, 对物种CH3的脱附或进一步转化为附加值高的化学品有利. 对TM-碳18环间的作用进行分析发现, 碳18环通过与TM的-共轭, 呈现良好的储存和吸电子的作用, 进而可以促进甲烷C—H活化能垒的降低. 碳18环基TM单原子具有温和条件下高效活化甲烷C—H的潜力.
甲烷; C—H活化; 碳18环; 单原子; 密度泛函理论
随着化石能源煤、 石油的消耗和化工生产伴随的大量二氧化碳气体排放所造成的全球气候恶化, 天然气、 页岩气和可燃冰的主要成分甲烷的转化和利用, 因其良好的氢碳比(4∶1)而受到极大关注[1~3]. 然而, 由于甲烷分子具有3杂化的高度对称的四面体结构, C—H键能非常高(435 kJ/mol), 远高于目标产物的C—H键能, 故其转化常需在高温高压等苛刻条件下进行[4,5], 因而容易发生深度转化, 无法合成具有高附加值的化学品. 如甲烷氧化偶联(OCM)反应在高温条件下生成的C2烃, 由于反应放热量大易转化为CO2[6]; 而现有的甲烷无氧转化路线需在高温条件下进行, 易发生积炭而导致催化剂失活[7]. 因此, 寻找高效稳定的催化剂, 提高甲烷C—H活化能力, 并促使甲烷在温和条件下的高效转化具有重要的意义.
近年来, 碳化硅或碳材料限域的过渡金属催化剂在应用于甲烷定向转化上呈现出了良好的前景. 如Guo等[8]发现碳化物限域的铁催化剂(FeSiC2)在反应温度1363 K下可使甲烷直接裂解, 转化为乙烯、 芳烃和氢气等高值化学品, 烃选择性高达99%, 他们对催化剂的表征和采用的密度泛函理论(DFT)的模拟揭示了配位不饱和的单原子Fe能有效抑制甲烷深度脱氢, 避免积炭; 而Cui等[9]发现石墨烯限域单原子Fe可在室温条件下直接催化甲烷转化为高附加值的C1含氧化合物, 并认为甲烷高效活化的原因在于限域的单中心Fe分解双氧水产生O—Fe—O活性物种, 吸电子基团O能显著降低甲烷活化的温度. 另一方面, Wu等[10]比较研究了11种不同石墨烯负载的单原子Fe对甲烷的活化作用, 发现具有单空位的石墨烯要比双空位石墨烯负载的单原子Fe具有更低的裂解甲烷第一C—H的能垒, 说明碳材料的结构会显著影响金属对甲烷C—H的活化能垒. 因此, 有必要深入探究这类催化剂的作用机制.
单原子催化剂具有原子利用率高、 电子结构易调控等优点[11~13], 而碳材料以其成本低、 耐高温和活性金属相易回收等特点成为金属单原子的优良配体[14,15]. 鉴于过渡金属Os和Ir单原子在甲烷C—H活化上的优异表现[16~20], 以及碳18环(C18)这一新近合成的一维碳材料所具有的特殊结构[21,22](长短 C—C交替循环、 环平面内与环平面外两套不同的离域大键), 本文选择考察过渡金属TM(Os, Ir)单原子键合在C18环上(TMC18)后对甲烷C—H的活化, 以理解TMC18的作用机制, 以及C18环在甲烷C—H活化中所起的作用.
1 计算方法
采用Gaussian 16程序[23]B97XD泛函[24]、 def2-TZVP基组[25]优化几何结构(其中, Os和Ir金属原子使用考虑了标量相对论效应的Stuttgart赝势), 原子间色散力校正则采用该泛函自带的DFT-D2[26]色散参数; 在此基础上, 采用B97XD/def2-QZVP或UCCSD(T)[27]/def2-QZVP[25]进行单点能计算以提高能量值的精度. 已有研究证实, 带长程校正的B97XD泛函可以正确预测C18环的几何结构和电子结构[22], 同时在含过渡金属(Au, Pt, Rh, Ir)的催化反应的能垒计算中B97XD泛函表现良好[28,29]; Tian等[30]考察了27个DFT泛函预测甲烷C—H活化的表观能垒, 发现采用B97XD泛函的DFT计算与公认的“黄金标准”稳健理论CCSD(T)/CBS计算提供的结果相当接近.
势能面上的稳态和过渡态结构均通过振动频率分析(Freq)有无虚频确定, 过渡态结构有且仅有一个虚频. 内禀反应坐标(IRC)计算确保过渡态连接反应路径中正确的反应物和产物. 所有几何结构的波函数均通过stable=opt检查以确认其稳定性. 电荷分析及轨道分析分别采用Hirshfeld[31]方法和NBO[32]程序完成. Wiberg键级采用多功能波函数分析程序Multiwfn 3.8(dev)[33]计算得到. 体系的自旋多重度以数字标注于相应物种的左上角.
甲烷加合物IM1生成的吸附自由能(ads)和甲基的脱附自由能(des)的计算如下:
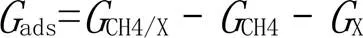

式中: X表示TM(Os, Ir), TM+(Os+, Ir+)或TMC18(TM = Os, Ir).
本文涉及的能量值均已包括零点能(ZPE), 且是在298 K, 1×105Pa条件下获得.
2 结果与讨论
2.1 OsC18和IrC18的结构与性能
图1为计算优化的C18环的几何结构. 可以看出, C18环是长短C—C(分别为0.135和0.122 nm)交替循环变化的9h对称性结构分子. 这与AFM/STM实验观察到的键长交替特征一致[21], 也与Liu等[22]采用高级别方法CCSD优化后得到的键长和点群对称性相同. 相比于具有垂直于2杂化碳环平面的离域键的石墨烯(C—C键长为0.142 nm)[34],杂化形成的C18环具有相互垂直的、 位于环平面内外的两套离域键以及C—C键呈类单-三键交替的结构特征.
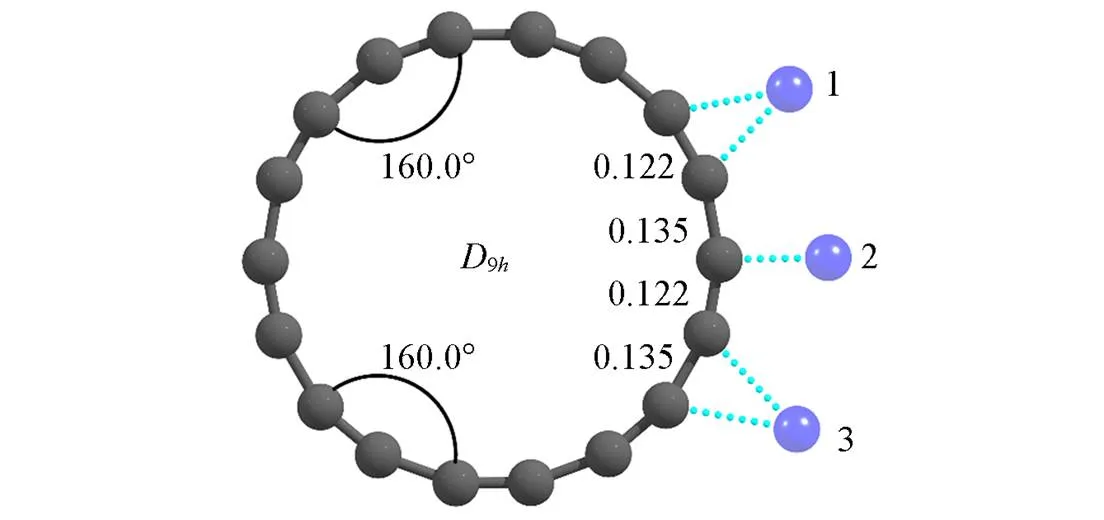
Fig.1 Optimized structures of cyclo[18] carbon(C18) at ωB97XD/def2⁃TZVP level and three possible sites for TM(Os, Ir) binding to cyclo[18] carbon
Bond lengths are in nm.
过渡金属原子Os和Ir与C18环有3个可能的结合位点, C—C短键、 C—C长键、 单个C原子(图1). 通过几何优化, 可以确定单原子Os和Ir键合于C18环上的最优位点: (1) Os单原子从C18环外优先桥连键合于其C—C短键; (2) 二重态和四重态Ir从C18环外桥连键合于其C—C短键, 而六重态Ir则从C18环外键合于其单个C原子. Vadalkar等[35]采用密度泛函理论在B97XD/6-311++G(,)水平下考察Al, Si和P原子与C18环结合时得到了相似的结果.
图2为计算优化的不同自旋的TMC18的几何结构. C18环的键长和键角会因Os, Ir与C18环的键合(原子间距分别为0.190~0.218 nm和0.190~0.214 nm)而发生改变. 桥连键合TM-(C—C短键)涉及的C—C键长由0.122 nm增至0.133~0.137 nm, 牵涉的C—C—C键角由160.0°缩小至约137.0°, 致使TMC18呈水滴型结构; 线连键合TM-C仅发生在6IrC18(六重态)上, 其相关C—C键长和C—C—C键角的变化类似.
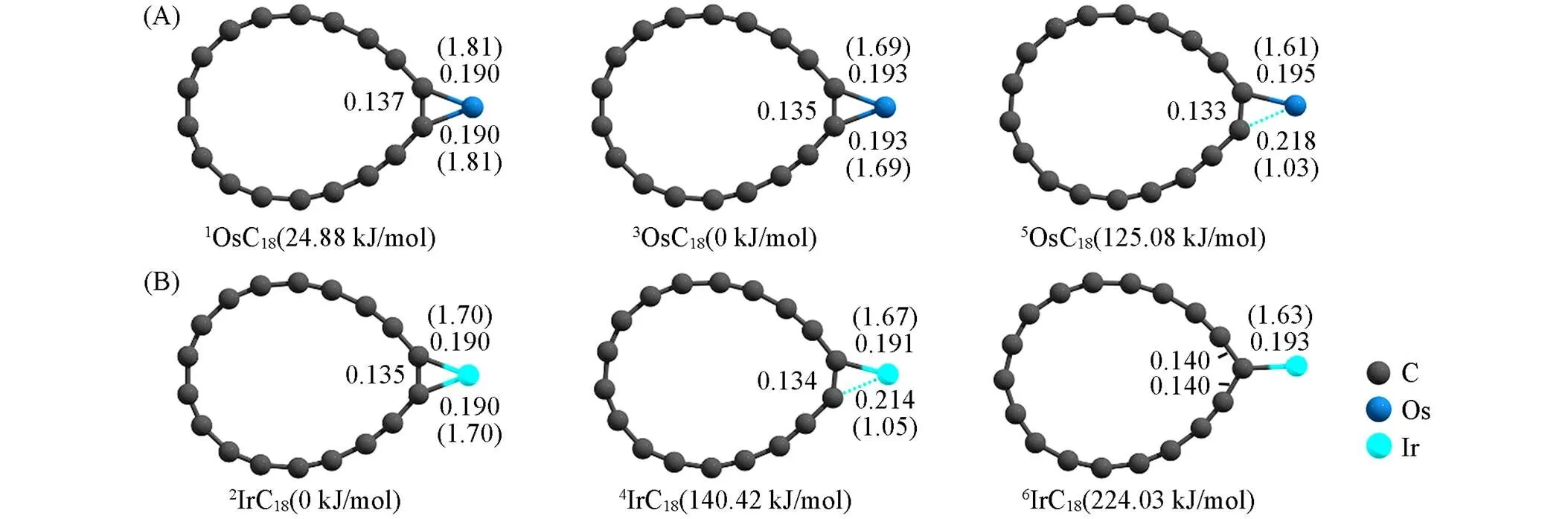
Fig.2 Predicted bond lengths(Wiberg bond order) of TM⁃C at ωB97XD/def2⁃TZVP level and total relative energies(ETOT) at ωB97XD/def2⁃QZVP level for OsC18 and IrC18 with different spin states
Bond lengths are in nm.
当自旋多重度由低向高递进时, TM桥连/线连键合C18的TM-C键长随之增长, 而相应的Wiberg键级则呈减小的趋势(表1), 从键级大小判断可以认为Os, Ir与C18环结合相对稳定. 与之相关的是,1OsC18(单重态)、3OsC18(三重态)和2IrC18(二重态)具有相对较低的总能量(TOT)(图2)和较高的TM-C18作用能(int)(表1), 即低自旋态的TMC18具有较高的稳定性, 同时int的正值反映出Os, Ir与C18环的结合在热力学上是有利的, 其中正值越大表示结合越稳定. 因此, 后续涉及TMC18的计算仅考虑OsC18的单重态、 三重态和IrC18的二重态.

Table 1 Bond lengths, Wiberg bond index(WBI) and the atomic orbital pair 5d(TM)-2p(C) contribution to WBI of TM-C and TM-C18 interaction energy(Eint)(TM=Os, Ir)
2.2 TM(Os, Ir)和TM+(Os+, Ir+)与CH4的相互作用
由于过渡金属TM与C18环结合, 从金属碳化物和电负性角度来看, TM带正电荷, 因此为了探究C18环对金属原子活化甲烷C—H的影响, 比较研究了金属和金属阳离子与甲烷的作用.
依据能量最低原则, 仅考虑5Os(五重态)和4Ir(四重态)参与活化甲烷的情况; 图3(A)给出了 TM(Os, Ir)单原子活化甲烷C—H的势能面(PESs), 势能面所涉关键物种的结构及参数如图4(A)所示. 活化过程为: TM与CH4作用形成加合物IM1, 然后TM插入到甲烷C—H, 使C—H断裂形成IM2, 过渡态TS为三中心结构, 与文献报道的采用B3LYP和BPW91两种不同泛函计算研究单原子Fe活化甲烷C—H的结果一致[36]. Os, Ir与甲烷作用生成加合物IM1的ads分别为7.53(10.42)和‒6.57(‒12.89) kJ/mol. Os, Ir活化甲烷C—H的自由能垒∆a分别为133.26(123.64)和103.72(74.94) kJ/mol, 反应自由能∆r分别为‒8.79(‒15.94)和‒8.58(‒16.19) kJ/mol. 括号内的数值为基于UCCSD(T)/def2-QZVP//B97XD/def2-TZVP水平的计算值, 与B97XD/def2-QZVP//B97XD/def2-TZVP水平计算结果相差不大; 已有文献[37]采用MPCF方法计算得到的五重态Os单原子对甲烷C—H的活化自由能垒和反应自由能分别为112.97和‒29.29 kJ/mol, 也与本文计算值接近. 这些说明了采用B97XD/def2-QZVP//B97XD/def2-TZVP进行计算是可行的.
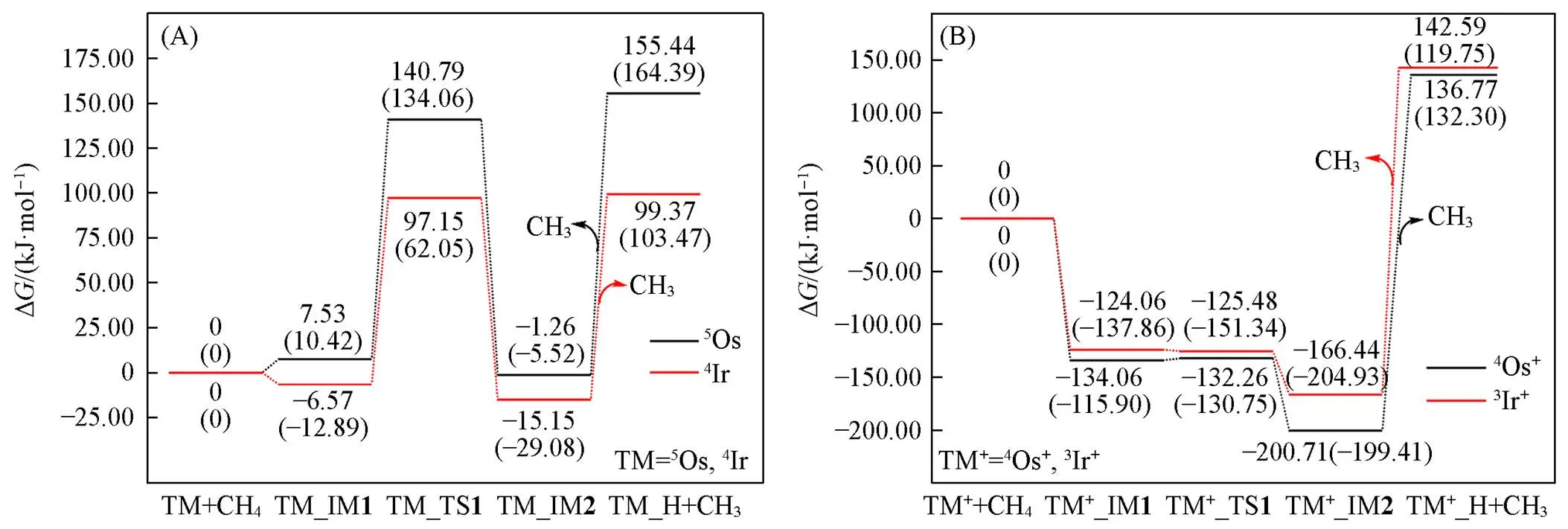
Fig.3 Predicted potential energy surfaces(PESs) for C—H activation of CH4 by TM(A) and TM+(B)(TM=Os, Ir) at the level of ωB97XD/def2⁃QZVP//ωB97XD/def2⁃TZVP and UCCSD(T)/def2⁃QZVP//ωB97XD/def2⁃TZVP(the latter shown in parenthesis)
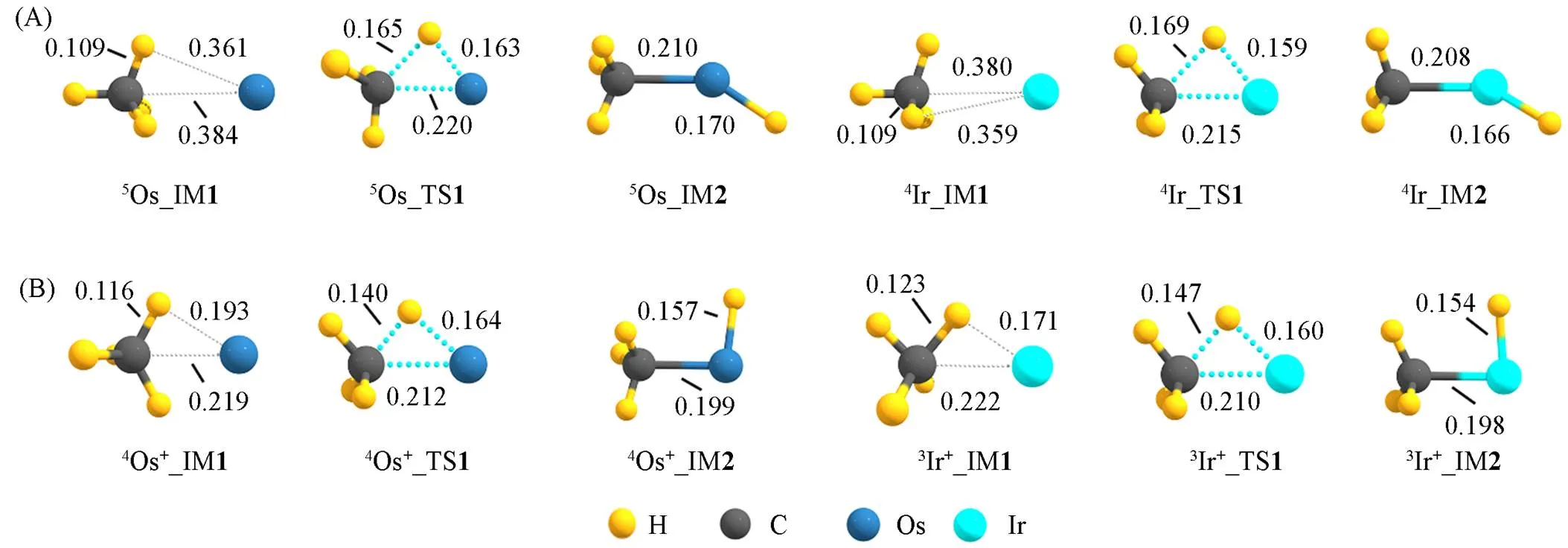
Fig.4 Geometric structures and parameters of the key species in PESs for C—H activation of CH4 by TM(A) and TM+(B)(TM=Os, Ir) at the level of ωB97XD/def2⁃TZVP
Bond lengths are in nm.
作为对比, 图3(B)给出了基态TM+(Os+, Ir+)对甲烷C—H活化的反应势能面, 势能面所涉及关键物种的结构及参数如图4(B)所示. 活化过程与TM对甲烷C—H活化类似, 即甲烷C—H活化经历加合物IM1生成、 三中心过渡态TS、 C—H键裂产物IM2生成. 所不同的是, 甲烷分子与TM+离子的作用相对更强. 如加合物IM1中Os+(Os)与甲烷C, H原子的间距分别为0.219(0.384)和0.193(0.361) nm; 甲烷预活化后的C—H键长为0.116(0.109) nm; 过渡态TS中Os+(Os)与甲烷C, H原子的间距分别为0.212(0.220)和0.164(0.163) nm; Ir+(Ir)的情况与此类似. 即相对于TM原子, 预活化过程中TM+离子与H—CH3分子碎片中C, H原子的间距较小, 预活化后的甲烷C—H键更长, 说明TM+与甲烷的作用较强. 而且Os+和Ir+形成甲烷加合物IM1的ads分别为‒134.06(‒115.90)和‒124.06(‒137.86) kJ/mol, 相比TM形成加合物在热力学上更为有利. 同时甲烷分子在Os+和Ir+上的活化自由能垒Δa分别为1.80 (‒14.85)和‒1.42(‒13.48) kJ/mol, 反应自由能Δr分别为‒66.65(‒83.51)和‒42.38(‒67.07) kJ/mol, 远低于Os和Ir原子活化甲烷分子的活化自由能垒和反应自由能. 说明TM+离子活化甲烷的热力学和动力学驱动力均远强于TM原子, 而Ir则强于Os. 这一观点得到Li等[17]和Armentrout等[19]采用DFT计算方法给出甲烷在Os+和Ir+阳离子上的第一C—H键裂是一自发过程的结果的佐证.
2.3 TMC18(TM=Os, Ir)与CH4的相互作用
图5给出了TMC18活化甲烷C—H关键物种的几何结构及参数, 甲烷C—H活化势能面如图6所示(自旋多重度依据最低能量原则确定), 活化过程为: 加合物IM1生成、 三中心过渡态TS、 C—H键裂产物IM2生成, TMC18与甲烷的作用强度介于TM和TM+之间. 以OsC18活化甲烷为例(IrC18的情况类似), IM1中活化的甲烷C, H原子与Os的最小间距分别为 0.234和0.208 nm, 过渡态TS中分别为0.209和0.168 nm, 甲烷预活化后的键长为 0.112 nm; 而Os+(Os)活化甲烷时, IM1中活化的甲烷C, H原子与Os+(Os)的最小间距分别为 0.219(0.384)和0.193(0.361) nm, 过渡态TS中分别为0.212(0.220)和0.164(0.163) nm, 甲烷预活化后的C—H键长的0.116(0.109) nm. 即TMC18预活化甲烷时, 甲烷C, H与TM间的距离介于TM, TM+预活化甲烷的情形之间.
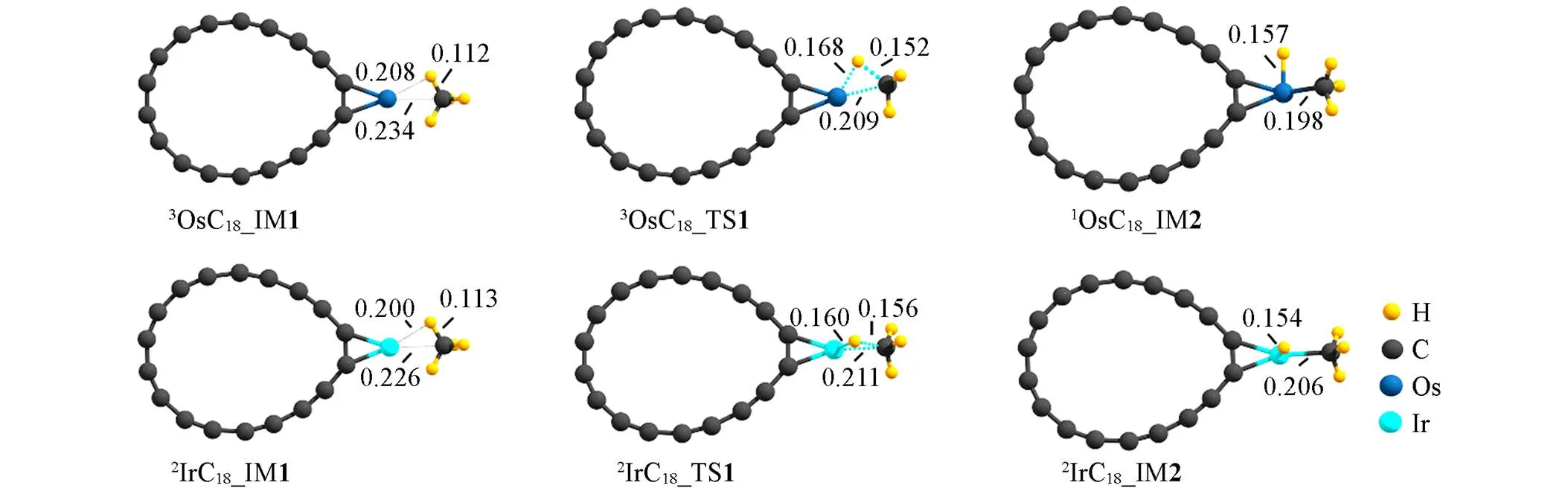
Fig.5 Geometric structures and parameters of the key species in PESs for C—H activation of CH4 by TMC18(TM=Os, Ir) at the level of ωB97XD/def2⁃TZVP
Bond lengths are in nm.
TMC18与甲烷间作用的强弱也可从能量学上进行分析. TMC18活化甲烷的活化自由能垒显著低于TM, 而略高于TM+. IrC18上甲烷C—H活化仅存在二重态途径; 而OsC18上存在单、 三重态途径, 因OsC18与甲烷作用生成的加合物的单重态能量略高于三重态, 甲烷C—H键裂产物的单重态能量却低于三重态[图6(A)], 故甲烷C—H活化势能面出现了自旋交叉的情形, 即三重态向单重态翻转. 由 图6(A)和(B)可知, 甲烷C—H在OsC18, IrC18上的活化自由能垒Δa分别为44.48和1.08 kJ/mol, 反应自由能∆r分别为‒23.22和‒17.12 kJ/mol; 而在Os(Os+), Ir(Ir+)上甲烷C—H活化自由能垒Δa分别为 133.26(1.80)和103.72(‒1.42) kJ/mol, 反应自由能Δr分别为‒8.79(‒66.65)和‒8.58(‒42.38) kJ/mol[图3(A)和(B)], 即甲烷C—H在TMC18(TM=Os, Ir)上的热力学和动力学驱动力介于TM和TM+之间.
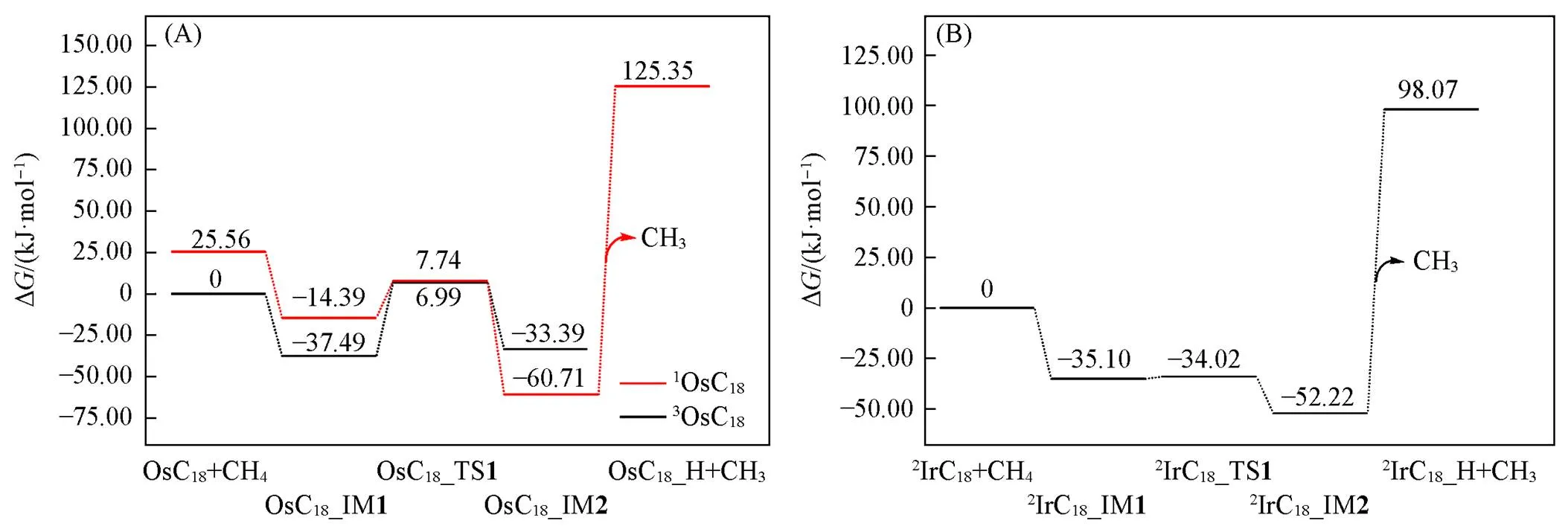
Fig.6 Predicted potential energy surfaces(PESs) for C—H activation of CH4 by OsC18(A) and IrC18(B) at the level of ωB97XD/def2⁃QZVP//ωB97XD/def2⁃TZVP
甲烷C—H在OsC18, IrC18上的活化自由能垒显著低于Os, Ir, 特别是IrC18, 表明C18环的存在使得甲烷C—H的活化变得容易; 另一方面, 甲烷C—H键裂产物(H+CH3)与Os+, Ir+的结合过强, CH3自其离开所需能量des分别为 337.48和309.03 kJ/mol[图3(A)和(B)], 远大于OsC18, IrC18上的186.06和150.29 kJ/mol[图6(A)和(B)], 说明甲烷C—H虽在TM+上易于活化, 但C—H键裂产物不易转化为高附加值化学品, 而会长时间停留在金属上发生连续脱氢、 形成积炭, 使催化剂失活; OsC18和IrC18则会在一定程度上避免这种情况的发生. Marcinkowski等[38]的DFT计算研究指出, CH中间体与Pt(111)的结合力度远大于Cu(111)是Pt催化剂相对容易结焦的原因; Li等[39]通过动力学实验和理论研究也得出, 锚定在MXene载体上的六方密堆积的纳米铂活化甲烷C—H时可以形成易脱附的CH3物种, 从而可有效抑制积炭的发生. 因此, TM不利于甲烷C—H活化, TM+利于甲烷C—H键活化, 但其产生的C—H键裂产物CH3物种倾向于深度脱氢而不利于后续偶联反应C2等高附加值产物的形成, 而与甲烷作用的强度处于二者之间的TMC18将会是一个比较有前景的甲烷选择活化催化剂.
2.4 电荷布居分析
表2列出了TM, TM+和TMC18活化甲烷C—H过程所涉及的Hirshfeld电荷布居的一些主要数据. TM活化甲烷过程中的加合物IM1、 过渡态TS中甲烷碎片电荷布居值(CH4)均为负, 说明TM活化甲烷过程中电子由TM流向甲烷(亲核活化); 而TM+和TMC18活化甲烷过程中的加合物IM1、 过渡态TS中甲烷碎片所带电荷布居值则为正, 说明TM+和TMC18活化甲烷过程中电子由甲烷流向TM(亲电活化), 转移的电子数越多, 甲烷C—H活化自由能垒Δa越低.
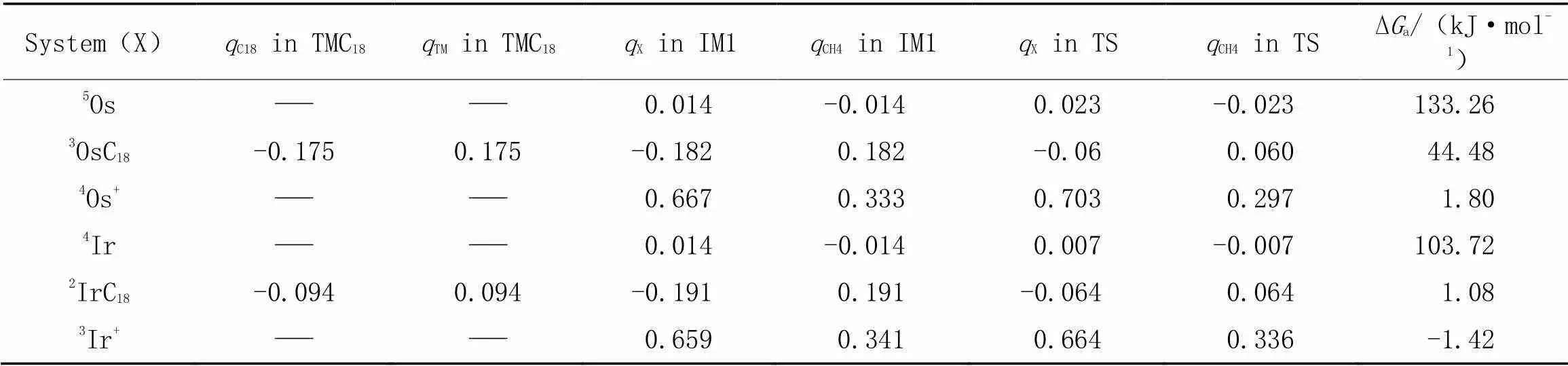
Table 2 Hirshfeld charge population(q) analysis on TMC18(TM=Os, Ir) and the adduct(IM1) and transition state(TS) involved into C—H activation of methane over TM, TM+ and TMC18, and the relevant activation free energy*
*qrepresentsTM,TM+orTMC18.
进一步比较TM, TM+, TMC18活化甲烷C—H的过渡态的电荷布居和活化能垒, 可知, TMC18能够降低其活化甲烷C—H能垒的原因是TM与C18环间相互作用导致部分电子从TM流向C18环, TMC18自身的电荷布居分析(TMC18中的C18为负)佐证了这一观点. 这与He等[40]认为羟基引入氧化铟团簇可以增大活性位点间的电荷差值从而降低其对CO2, CH4作用的活化自由能垒结论类似.
TMC18中TM和C18间的作用与其电荷转移程度有关. 图S1(见本文支持信息)采用原子着色的方式表示出了TMC18中的原子电荷分布: 正电荷(红色)主要分布在TM上, 负电荷(蓝色)主要分布在与TM连接的碳原子上. 表明C18环与C60一样, 是一个良好的吸电子载体[41]. 电荷转移加强了TM单原子与C18环之间的相互作用. 通过对描述TM-C18作用强弱的Wiberg键级进行分解分析, 发现TM的5轨道和键连C原子的2轨道间的相互作用对总键级的贡献约占60%(表1), 说明TM的5轨道与C原子的2轨道间存在很大的重叠, 5(TM)-2(C)轨道相互作用显著. 这种作用实际上就是Os和Ir的轨道与C18环轨道重叠形成的强-共轭(图7). 这种强的-共轭作用一方面促进TM单原子与C18环间的电荷转移, 使体系趋于稳定; 另一方面具有阻止TM单原子在碳18载体上的迁移与团聚的能力, 与Pantha等[42]认为Fe, Co, Ni金属原子与石墨烯的强结合源于金属轨道和石墨烯轨道的共价重叠观点一致.

Fig.7 Relevant NBO orbital interactions in TMC18(TM=Os, Ir)
3 结 论
采用密度泛函理论方法计算考察了最小全碳化合物C18环与过渡金属单原子Os和Ir的作用, 及其对甲烷C—H的活化. 结果表明, C18环表现为吸电子载体, 其与过渡金属单原子间较强的-共轭作用可以促进金属上的电子流向C18环, 进而可显著降低甲烷C—H的活化能垒. 比较过渡金属TM单原子、 单价阳离子以及TMC18三者活化甲烷C—H的行为, 发现虽然甲烷C—H活化的过渡态均具有三中心结构, 但其能量学却存在明显差异. TM单原子活化甲烷C—H的活化自由能垒极高, 以致常规条件下甲烷C—H活化难以进行; TM+上甲烷C—H活化却是一个可自发进行的过程, 但因C—H键裂产物与金属结合很强, 金属易发生积炭失活; 然而, TMC18活化甲烷C—H的活化自由能垒很低, 其C—H键裂产物与金属的结合强度也明显低于TM+. 因此, TMC18具有在温和条件下高效活化甲烷C—H的潜力.
支持信息见http: //www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20230259.
[1] McFarland E.,,2012,(6105), 340—342
[2] Höök M., Tang X.,,2013,, 797—809
[3] Meng X., Cui X., Rajan N. P., Yu L., Deng D., Bao X.,,2019,(9), 2296—2325
[4] Spivey J. J., Hutchings G.,,2014,(3), 792—803
[5] Kwon Y., Kim T. Y., Kwon G., Yi J., Lee H.,,2017,(48), 17694—17699
[6] Wang P., Zhao G., Wang Y., Lu Y.,,2017,(6), e1603180
[7] Huang Z. Q., Chen Y. T., Chang C. R., Li J.,,2021,(21), 13149—13159
[8] Guo X., Fang G., Li G., Ma H., Fan H., Yu L., Ma C., Wu X., Deng D., Wei M., Tan D., Si R., Zhang S., Li J., Sun L., Tang Z., Pan X., Bao X.,,2014,(6184), 616—619
[9] Cui X. J., Li H. B., Wang Y., Hu Y. L., Hua L., Li H. Y., Han X. W., Liu Q. F., Yang F., He L. M., Chen X. Q., Li Q. Y., Xiao J. P., Deng D. H., Bao X. H.,,2018,(8), 1902—1910
[10] Wu C. C., Gates I. D.,,2019,, 40—47
[11] Qiao B., Wang A., Yang X., Allard L. F., Jiang Z., Cui Y., Liu J., Li J., Zhang T.,,2011,(8), 634—641
[12] Liu K., Zhao X., Ren G., Yang T., Ren Y., Lee A. F., Su Y., Pan X., Zhang J., Chen Z., Yang J., Liu X., Zhou T., Xi W., Luo J., Zeng C., Matsumoto H., Liu W., Jiang Q., Wilson K., Wang A., Qiao B., Li W., Zhang T.,,2020,(1), 1263
[13] Zhang T., Wang A., Liu H., Liu X.,,2021,(2), 175—187
[14] Gerber I. C., Serp P.,,2020,(2), 1250—1349
[15] Zhang J., Li X., Chen H., Qi M., Zhang G., Hu H., Ma X.,,2017,(31), 19755—19775
[16] Armentrout P. B.,,2017,(1), 10—18
[17] Li F. X., Zhang X. G., Armentrout P. B.,,2006,, 279—300
[18] Zhang G. B., Li S. H., Jiang Y. S.,,2003,(19), 3820—3830
[19] Armentrout P. B., Parke L., Hinton C., Citir M.,,2013,(9), 1157—1173
[20] Li W., Wu X., Liu Z., Wu H., Zhang D., Ding X.,,2020,(19), 8346—8351
[21] Kaiser K., Scriven L. M., Schulz F., Gawel P., Gross L., Anderson H. L.,,2019,(6459), 1299—1301
[22] Liu Z. Y., Lu T., Chen Q. X.,,2020,, 468—475
[23] Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams⁃Young D., Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., Fox D. J.,16,.03, Gaussian Inc., Wallingford CT, 2016
[24] Chai J. D., Head⁃Gordon M.,,2008,(44), 6615—6620
[25] Weigend F., Ahlrichs R.,,2005,(18), 3297—3305
[26] Grimme S.,,2006,(15), 1787—1799
[27] Purvis G. D., Bartlett R. J.,,1982,(4), 1910—1918
[28] Carmona M., Perez R., Ferrer J., Rodriguez R., Passarelli V., Lahoz F. J., Garcia⁃Orduna P., Carmona D.,,2022,(33), 13149—13164
[29] Kang R., Lai W., Yao J., Shaik S., Chen H.,,2012,(9), 3119—3127
[30] Tian T., Sun X., Weiske T., Cai Y., Geng C., Li J., Schwarz H.,,2019,(14), 1812—1821
[31] Hirshfeld F. L.,,1977,(2), 129—138
[32] Glendening E., Badenhoop J., Reed A., Carpenter J., Weinhold F.,, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 1996
[33] Lu T., Chen F.,,2012,(5), 580—592
[34] Yang G., Li L., Lee W. B., Ng M. C.,,2018,(1), 613—648
[35] Vadalkar S., Chodvadiya D., Vyas K. N., Jha P. K.,,2022,, 229—237
[36] Sun Q., Li Z., Du A., Chen J., Zhu Z., Smith S. C.,,2012,(1), 291—297
[37] Swang O., Faegri K., Gropen O.,,2002,(11), 3006—3009
[38] Marcinkowski M. D., Darby M. T., Liu J., Wimble J. M., Lucci F. R., Lee S., Michaelides A., Flytzani⁃Stephanopoulos M., Stamatakis M., Sykes E. C. H.,,2018,(3), 325—332
[39] Li Z., Xiao Y., Chowdhury P. R., Wu Z., Ma T., Chen J. Z., Wan G., Kim T. H., Jing D., He P., Potdar P. J., Zhou L., Zeng Z., Ruan X., Miller J. T., Greeley J. P., Wu Y., Varma A.,,2021,(10), 882—891
[40] He H. R., Xia W. S., Zhang Q. H., Wan H. L.,, 2022,(8), 20220196(何鸿锐, 夏文生, 张庆红, 万惠霖. 高等学校化学学报,2022,(8), 20220196)
[41] Stasyuk A. J., Stasyuk O. A., Sola M., Voityuk A. A.,.(),2020,(3), 352—355
[42] Pantha N., Ulman K., Narasimhan S.,,2020,(24), 244701
Methane C—H Activation by Cyclo[18] Carbon-based Single-atom Transition Metal(Os, Ir)
ZHANGHaiping, KONGXue, XIAWensheng*, ZHANGQinghong, WANHuilin
(,,,,,361005,)
It is of great significance and challenge for the conversion and utilization of methane(the main component of natural gases) due to its high stability in chemistry. In order to breakthrough out it, the key is to develop the catalysts for methane C—H activation under mild conditions. Here, we investigated methane C—H activation over cyclo[18] carbon-based single-atom transition metal(TM=Os, Ir)(TMC18) by means of density functional theory(DFT). The results show that the activation barrier of methane C—H over TMC18is significantly lower than TM itself, and the species CH3resulted from methane C—H cleavage tends to bind more weakly with TMC18than TM+ions. The weakened interaction between CH3and TMC18favors to the CH3desorption or further transformation into value-added chemicals. Thus, cyclo[18] carbon-based single-atom TM exhibits its promising ability to high-efficiently activate C—H of methane at mild conditions. The detailed analysis on the interaction of TM with cyclo[18] carbon indicate that cyclo[18] carbon is good in electron-storage/attraction by-conjugation of TM-cyclo[18] carbon, which leads to the decrease in barrier of methane C—H activation over cyclo[18] carbon-based TM.
Methane; C—H activation; Cyclo[18] carbon; Single atom; Density functional theory
2023-05-30
夏文生, 男, 博士, 教授, 主要从事催化和理论化学研究. E-mail: wsxia@xmu.edu.cn
国家重点研发计划“政府间国际科技创新合作/港澳台科技创新合作”重点专项项目(批准号: 2019YFE04400)和教育部创新团队发展计划项目(批准号: IRT1036)资助.
O643
A
10.7503/cjcu20230259
2023-08-09.
Supported by the National Key Research Plan “Intergovernmental International Innovation of Science and Technology Cooperation/Hong Kong, Macao and Taiwan Technological Cooperation” Project, China(No.2019YFE04400) and the Ministry of Education Innovation Team Development Program Project, China(No.IRT1036).
(Ed.: Y, K, S)
猜你喜欢
食品工业科技(2022年22期)2022-11-11
北京航空航天大学学报(2022年5期)2022-06-06
药学研究(2022年12期)2022-02-04
大学化学(2021年8期)2021-09-26
食品与发酵工业(2021年14期)2021-08-02
中学课程辅导·教学研究(2021年8期)2021-07-14
化学工业与工程(2021年3期)2021-06-25
燃料化学学报(2021年5期)2021-06-02
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08