
误诊为先天性肾上腺皮质增生症的新生儿假性醛固酮减少症一例
2023-12-11 03:12赵婉君
中国临床新医学 2023年11期
赵婉君, 张 娟
作者单位:100730 北京,北京医院·国家老年医学中心·中国医学科学院老年医学研究院新生儿科(赵婉君);100191 北京,北京大学第三医院儿科(张 娟)
1 病例介绍
患儿,女,因“呼吸困难10 min”于2021年5月7日收住北京大学第三医院儿科,系第2胎第2产,胎龄32+4周,试管婴儿,因“双胎妊娠、母亲重度子痫前期”剖宫产娩出,无宫内窘迫及生后窒息,出生体重1 740 g。父母体健,否认遗传性疾病家族史。双胎之大男,体重1 560 g,无特殊并发症。患儿入院查体无异常,经呼吸支持、营养支持后第9天达全肠内喂养。生后13 d时监测血钠(128 mmol/L)、血氯(94 mmol/L)明显低于正常值,血钾(6.28 mmol/L)明显高于正常值(见图1),同期血气分析提示代谢性酸中毒(pH 7.30,BE-7 mmol/L)。甲状腺功能无异常,肝肾功能、心肌酶在正常范围内,腹部超声提示双肾未见异常,头颅超声未见异常。应用沙丁胺醇雾化促进钾离子排出,静脉补钠治疗[3 mmol/(kg·d)]。患儿出生后14~23 d监测血钠低,波动在119.9~124.0 mmol/L;血钾偏高,波动在4.86~5.84 mmol/L,继续增加补钠量[静脉及口服共合6 mmol/(kg·d)]后,监测血钠波动在129~132 mmol/L。患儿于出生后16 d内分泌激素检查示:晨起促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)29.6 pg/ml正常;皮质醇(5.6 μg/dl)正常;17-羟孕酮(996 ng/dl)升高(参考范围26~568 ng/dl);睾酮(<0.69 nmol/L)正常,雄烯二酮(13.8 nmol/L)偏高(参考范围2.2~4.2 nmol/L)。生后20 d肾素活性[>12 ng/(ml·h)]升高[参考范围3~6 ng/(ml·h)],血管紧张素Ⅱ(>800 pg/ml)升高,醛固酮(1 859.9 pg/ml)升高(参考范围190~1 410 pg/ml)。生后29 d ACTH激发试验:静脉给予ACTH 0.25 mg/m2,17-羟孕酮(基础值)431 ng/dl,30 min值2 467 ng/dl,60 min值1 460 ng/dl。根据患儿存在的低钠血症、高钾血症、代谢性酸中毒,且雄激素水平偏高,ACTH激发试验值为1 000~10 000 ng/dl,临床初步诊断为不典型先天性肾上腺皮质增生症(congenital adrenal hyperplasia,CAH),并应用氢化可的松[10 mg/(m2·d)]口服治疗。采集患儿外周血样本进行全外显子测序分析,结果提示患儿外周血核型分析未见异常,基因拷贝数测序(copy number variation,CNV)检测结果提示4q31.23位置(chr4:148968002-149035453)存在杂合缺失,变异区域涵盖基因包括NR3C2基因8~9号外显子缺失。NR3C2基因突变引起可编码的盐皮质激素受体失活,造成醛固酮抵抗,从而出现失盐症状。该区域缺失变异在人类基因突变数据库尚未见报道。根据患儿临床表现及基因测序结果,修正诊断为假性醛固酮减少症Ⅰ型(pseudohypoaldosteronism typeⅠ,PHAⅠ),予停用氢化可的松,继续口服生理盐水补钠治疗至4月龄时监测血清钠持续在正常范围。患儿11月龄时监测体重8 kg,身长72 cm,均在同年龄同性别儿童第10百分位。
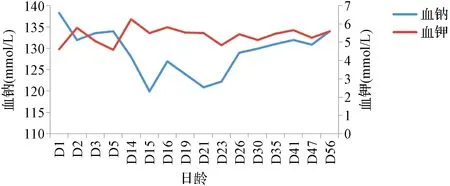
图1 患儿血钠、血钾值变化折线图
2 讨论
2.1本例患儿在新生儿期出现严重电解质紊乱,表现为低钠血症、高钾血症及代谢性酸中毒,血醛固酮及肾素水平升高,但监测雄激素值及17-羟孕酮值轻度升高,应用糖皮质激素治疗效果不佳,持续纠正电解质紊乱治疗至4月龄实验室指标恢复正常。结合患儿的临床表现、治疗效果及基因检测,最终诊断为PHAⅠ。
2.2PHA是新生儿或婴儿期盐皮质激素受体失活,细胞内醛固酮信号通路障碍,出现低钠血症、高钾血症、高醛固酮血症为主要表现的罕见疾病[1],最早由Cheek和Perry[2]于1958年报道。PHA患儿早期生化检测常难以与CAH进行区分,临床上易误诊、误治。CAH是肾上腺皮质激素合成过程中所需酶缺陷,继而引起皮质醇激素合成不足,进而引起前体物质分泌过多继发高雄激素血症的症候群。因新生儿酶缺乏程度差异较大,故临床代谢紊乱及疾病严重程度个体差异也较大,尤其是不典型CAH患儿,根据17-羟孕酮值诊断具有不确定性[3]。一项研究显示,胎龄、体重及采样时间可影响患儿17-羟孕酮值,胎龄32~36周,生后超过7 d早产儿17-羟孕酮(基础值)截点为1 206 ng/dl[4]。早产儿、低出生体重儿,由于肾上腺的发育不成熟,21-羟化酶、11-羟化酶延迟表达,17-羟孕酮浓度常常偏高,导致假阳性结果。本例患儿为早产儿,雄激素及17-羟孕酮值偏高导致误诊为CAH。临床上患儿无明显色素沉着或男性化表现等符合CAH的临床表征,临床医师应更关注患儿升高的醛固酮及肾素值。PHA患者血清醛固酮及肾素水平会明显升高[5]。CAH患儿应用糖皮质激素治疗效果显著,借此也可进一步鉴别PHA和CHA。PHA除了明显的高醛固酮血症外,也有研究提出可应用肾小管钾离子浓度梯度(transtubular potassium concentration gradient,TTKG)来反映潜在的盐皮质抵抗或缺乏,通过测定血/尿钾浓度和渗透压比值来计算。正常喂养下,TTKG通常为8~9,低TTKG值提示可能存在醛固酮抵抗或减少[6]。当患儿出现严重的失盐表现时,临床医师需结合患儿临床表现及多种激素水平进行分析,同时要注意有无性别、胎龄、日龄等多种因素的影响,早期基因诊断十分重要。
2.3本例患儿通过基因检测证实存在NR3C2基因8~9外显子杂合缺失变异。人类盐皮质激素受体由NR3C2基因编码,位于4q31.1与4q31.2之间,包括10个外显子,其中外显子8和外显子9的第一部分编码配体结合结构域[7]。Geller等[8]在1998年报道常染色体显性遗传PHAⅠ是由NR3C2基因突变引起编码的盐皮质激素受体失活,从而造成醛固酮结合障碍,钠通道表达减少。本病罕见,据英国一项数据统计其发病率为1/66 000[9]。近年来,人类基因突变数据库报道了60余种NR3C2基因的突变,包括错义突变、无义突变、剪切位点突变、缺失、插入及大片段的缺失[10],大多是不同的单核苷酸变异,像本例患儿的基因拷贝数变异的报道较少。本病临床上仅表现为肾小管对醛固酮抵抗,亦称为肾性PHAⅠ,病情相对较轻,临床表现个体差异较大,症状出现在新生儿期常有耗盐表现,可伴有生长发育迟缓,随年龄增长,通常能够在3岁前停止治疗,长期并发症较少,在成人期甚至无明显症状出现。但也有研究报道本病与新生儿期高病死率有关[11-12]。本例患儿在新生儿期发生严重电解质紊乱,经积极对症治疗,4月龄时实验室指标恢复正常,后期随访至11月龄达正常儿童生长发育水平。本例不足之处是未能进一步验证其父母基因以证实其遗传方式。
综上,本例患儿在新生儿期出现不明原因低钠血症、高钾血症及代谢性酸中毒,通过基因诊断发现了NR3C2基因新型缺失变异,确诊为罕见的PHAⅠ。笔者进行了与CAH的鉴别,拓宽了与PHA相关基因突变的遗传谱,有利于提高儿科临床医师对此类疾病的认识和鉴别诊断。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国生殖健康(2020年4期)2021-01-18
心肺血管病杂志(2019年12期)2019-05-20
中国生殖健康(2018年4期)2018-11-06
当代医学(2018年14期)2018-05-19
中国继续医学教育(2016年18期)2016-02-15
淮海医药(2015年2期)2016-01-12
现代检验医学杂志(2015年5期)2015-02-06
湖北农业科学(2014年11期)2014-09-10
