
煤自燃过程中H2来源理论研究
2023-12-16 08:01崔崇斌孙丽荣李伟刚郝朝瑜
山西焦煤科技 2023年11期
崔崇斌,孙丽荣,郑 立,李伟刚,郝朝瑜
(1.西山煤电(集团)有限责任公司 马兰矿,山西 太原 030205;2.太原理工大学 安全与应急管理工程学院,山西 太原 030024)
煤自燃是影响煤矿安全生产的主要危害之一,其不仅造成煤炭资源的极大浪费,而且还会引起瓦斯和煤尘爆炸等次生灾害。煤自燃指标气体探测法具有操作简易、准确度高、不干扰生产等优点,广泛应用于矿井防火实际中。然而,目前大多数研究专注于碳氧化合物、饱和烃和不饱和烃等气体,对H2的考量较少[1-3]. 目前关于H2作为煤自燃评价指标气体和其来源研究尚处于理论阶段。Samuel L.等[4]指出烟煤在55~95 ℃时便会产生可以观测到的H2,且其产生量与温度呈正相关。Wang等[5]指出醛类化合物是H2的一个明显来源,并提出了一条煤的低温氧化过程中形成H2的途径。王娅[6]提出复合指标气体浓度概念,在以往碳氧化合物、饱和烃和不饱和烃等气体之外,引入了对H2的考量,提高了指标气体预测范围。韩亮[7]指出随着煤化度的升高,煤中所含氢元素逐渐减少,H2的生成量也逐渐减小,生成速率也相应下降。姚彦娜[8]指出H2释放量随煤温均呈良好的指数规律,符合指标气体的选取原则。由此可见,对于H2研究主要局限于其作为特征气体,且主要通过实验方法实现,而缺少对H2来源理论层面的分析。
煤自燃是典型的自由基参与的反应[9,10]. H·作为最普遍的自由基,对煤自燃过程有重要影响,特别是自燃过程中特征气体的释放[11]. 因此,本文将H·作为反应初始点,分析不同位点H与H·的反应差异,并指出煤自燃过程中的H2释放机理,为井下防灭火工作提供一定理论指导。
1 材料与方法
1.1 计算模型
为保证效率和尽可能分析多种H位点,采用多种小模型来模拟不同位点,见图1(模型来源参考各类成熟模型[12,13]). 这类简化模型既节约了计算成本也充分考虑了多种条件,在微观模拟中非常普遍[14].
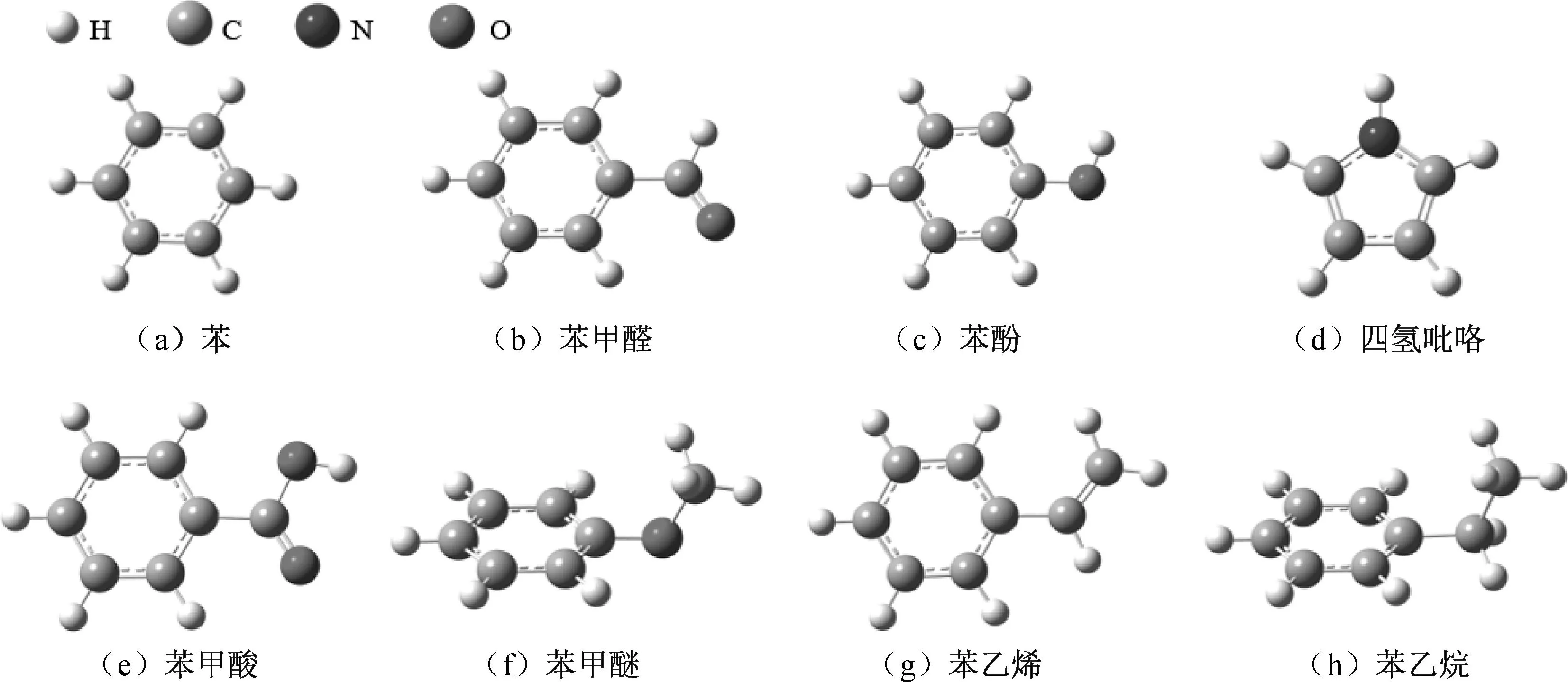
图1 模型图
1.2 计算方法
所有结构和能量计算都采用B3LYP计算方法,基组采用6-311 G(d,p). 热力学计算参数设定温度为298.150 K,压力为1 Atm(即常温常压)。所有能量计算均考虑零点能。
解离能[15](Bond dissociation energy,BDE)计算公式如下:
EBDE=Ea+b-Ea-Eb
(1)
式中:Ea+b为模型总能量,kJ·mol-1;Ea、Eb为分子片段能量(单独优化后),kJ·mol-1.
解离能反映不同位点H原子的断键所需要的能量,其数值大小可以反映出H原子参与反应的难易程度,数值越大,说明此处H原子越稳定,难以参与反应,反之同理。
反应平衡常数[16]计算公式如下:
(2)
式中:ΔG为反应自由能变,J·mol-1;R为气体摩尔常数,J·(K·mol)-1,取8.314;T为绝对温度,K.
反应平衡常数是描述反应进行程度的物理量,通常以105为界限,超过视为完全反应,低于10-5为视为完全不反应,介于两者之间表示反应不完全。
2 结果与讨论
2.1 前线轨道分析
前线轨道采用波函数分析软件Multiwfn[17]计算,出图采用分子可视化软件VMD[18]导出。
计算所得不同模型前线轨道分析见表1. 考虑部分模型对称性较高,可能存在轨道简并,因而表1中列出LUMO+1、LUMO+2等轨道能级数据。
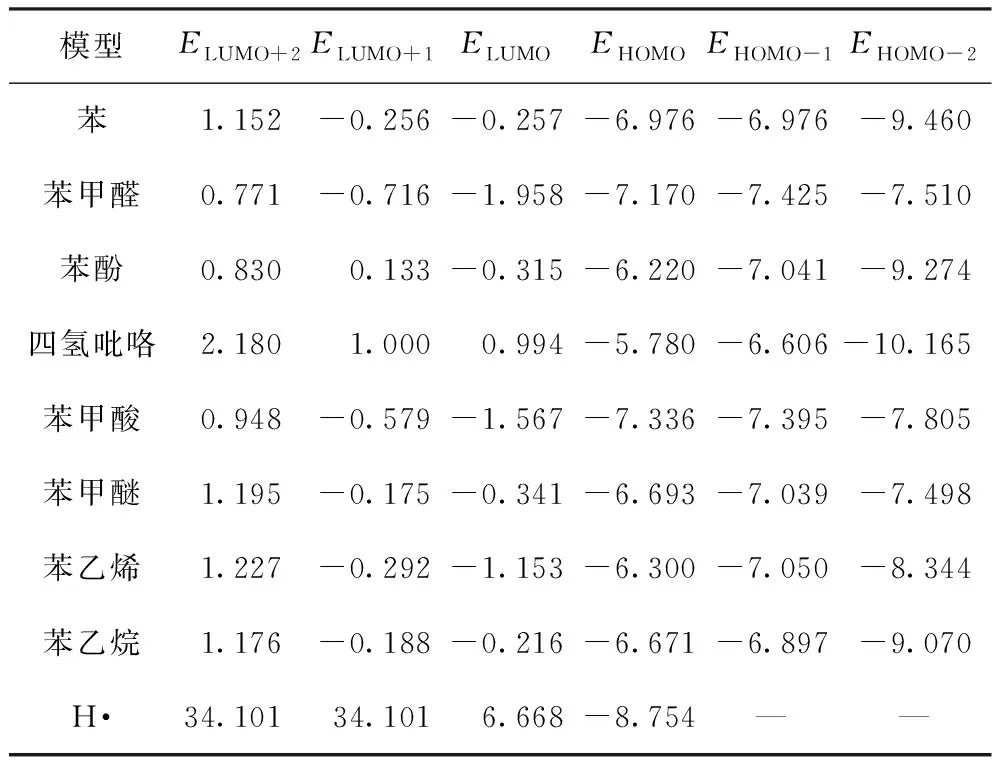
表1 不同模型前线轨道能级表 eV
由前线轨道理论可知,在分子的所有轨道中,能量最高的HOMO倾向于提供电子,而能量最低的LUMO更容易获得电子。反应物分子间前线轨道能级差越小,两者相互作用越强,反应倾向性越明显。本文采用前线轨道指出不同模型与H·反应的倾向性,从定性角度分析不同模型H2释放的差异。
由表1可知,模型苯HOMO与HOMO-1和四氢吡咯LUMO+1与LUMO存在轨道简并(这两种模型能极差较大,不可能主导H2的产生,省略具体分析),而其余模型均不存在轨道简并。经过简单计算比较可发现,上述模型与H·的最低能极差均为模型LUMO与H·的HOMO产生,能极差计算结果见表2,各模型LUMO见图2. 图中LUMO形状直观地反映了最高占据轨道上电子在各原子上的分布情况。

表2 不同模型与H·前线轨道能级差表
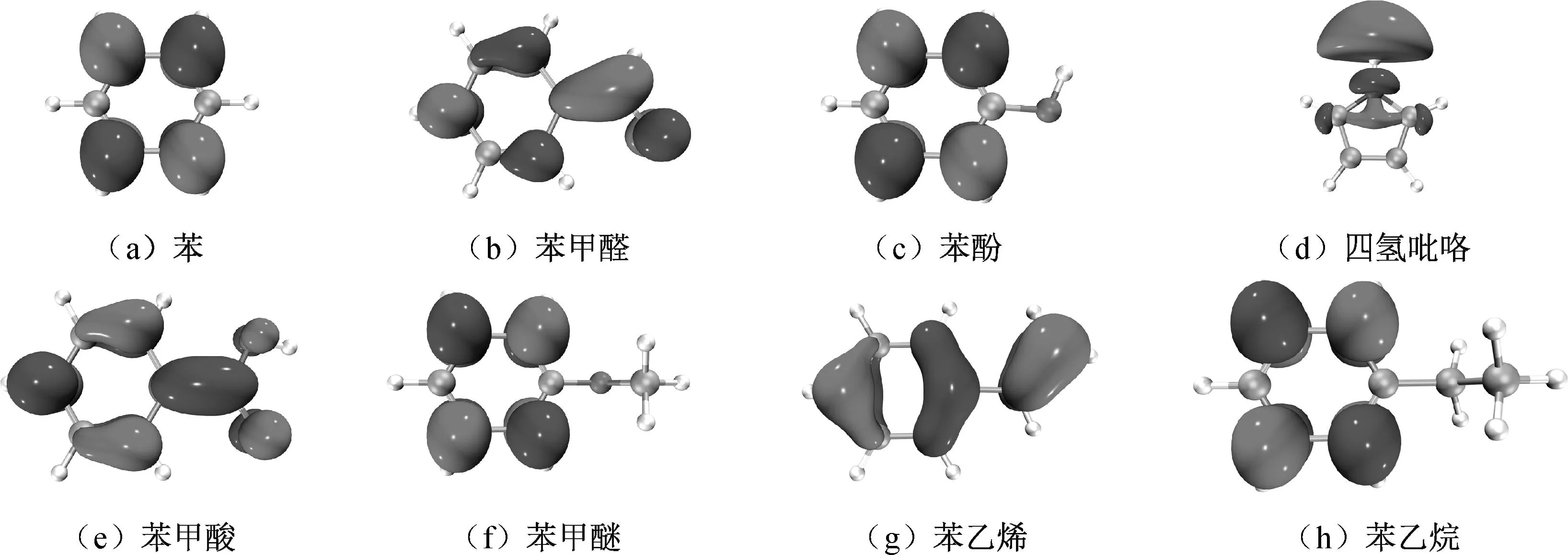
图2 不同模型LUMO图
由表2可知,根据能量相近原则,上述模型与H·的反应倾向性排序为:苯甲醛>苯甲酸>苯乙烯>苯甲醚>苯酚>苯>苯乙烷>四氢吡咯。显然,随着氧原子的引入,或者说煤变质程度的下降,模型与H·的反应倾向明显增强。
需要注意的是上述轨道分析仅指出与模型与H·的反应倾向,并未说明模型与H·的反应位点就是上图区域。以往研究表明,由于苯环这一高度共轭体系的存在,芳香环位点通常不易发生反应,通常还应分析具体位点轨道占据情况[19]. 考虑所有模型的轨道分析计算量较大,下文采用解离能针对具体位点展开直接分析。
2.2 不同位点H解离能
由图1可得,8种模型中存在多种H位点,但大多数位点化学环境相似(如苯环中与含氧官能团呈邻、间和对的H位点),经过筛选,在表3中仅保留差别较大的H位点。表中列出不同位点H的解离能与对应键长。另外,表头中四氢吡咯N、四氢吡咯C表示四氢吡咯中H原子所连接的原子类型(N或C).

表3 不同模型H位点解离能表
由表3可得,不同位点H解离能大小趋势为:四氢吡咯C>苯>苯乙烯>苯甲酸>苯乙烷>四氢吡咯N>苯甲醚>苯甲醛>苯酚。显然,与前文类似,随着氧原子的引入,模型中H位点解离能明显下降,从另一个角度说明与H·的反应更容易进行。
由于键长与具体的成键环境相关,将上述9种H位点归类为:烯H、烷H和其他H.
烯H包括:苯、苯乙烯、四氢吡咯C. 烯H解离能在所有位点中最大,而这部分也是煤结构中的主体,即典型芳香环结构。苯与苯乙烯中H位点键长一致,解离能几乎相等。而四氢吡咯C位点,键长收缩,原子吸引变强,解离能增大。
烷H包括:苯乙烷、苯甲醚。烷H解离能处于中间大小,这部分主要以芳香环侧链等形式存在于煤中。苯甲醚中C原子电子部分转移到O上,导致对H的吸引减弱,键长伸长。
其他H包括:四氢吡咯N、苯酚、苯甲酸、苯甲醛。其他H的解离能大小无明显规律,但整体上处于中下区间,其成因与具体位点相关。以苯酚为例,虽然此时H受到O原子的吸引作用,但苯酚中羟基直接与苯环相连,富含电子的O原子向苯环(π电子)区域传递电子,进而造成对H的吸引减弱;而与之对应苯甲酸中O原子直接连接在C原子上,其电负性更强,导致电子向O原子集中,H解离能明显增大。
综上所述,随着煤变质程度的降低,非碳元素含量升高,煤在自燃过程中更易产生H2.
2.3 反应热力学分析
前文分析仅指出了反应的倾向性和难易程度,缺乏针对具体反应的热力学层面分析,不能指出各反应极限。下文拟结合热力学分析,对反应物和产物展开分析,并计算反应平衡常数。
根据前文模型,列出如图3所示的反应方程式。此外,由于四氢吡咯C位点的解离能明显大于四氢吡咯N,故省略对四氢吡咯C的分析。反应热力学分析表见表4.
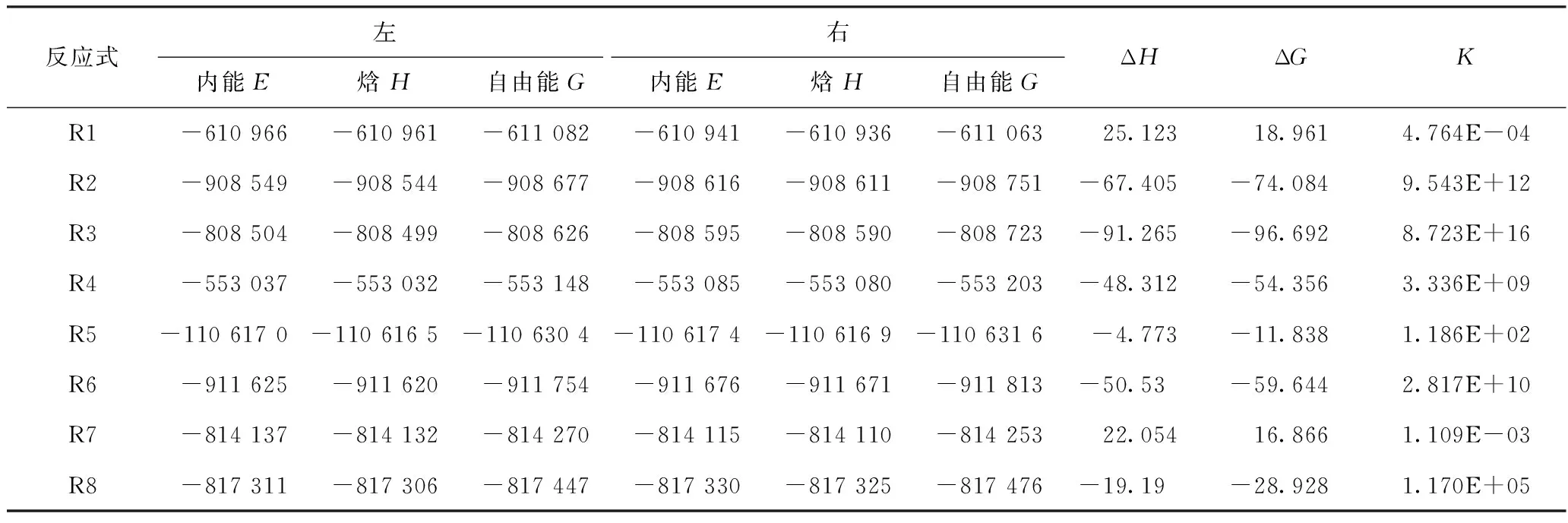
表4 反应热力学分析表(kJ·mol-1)
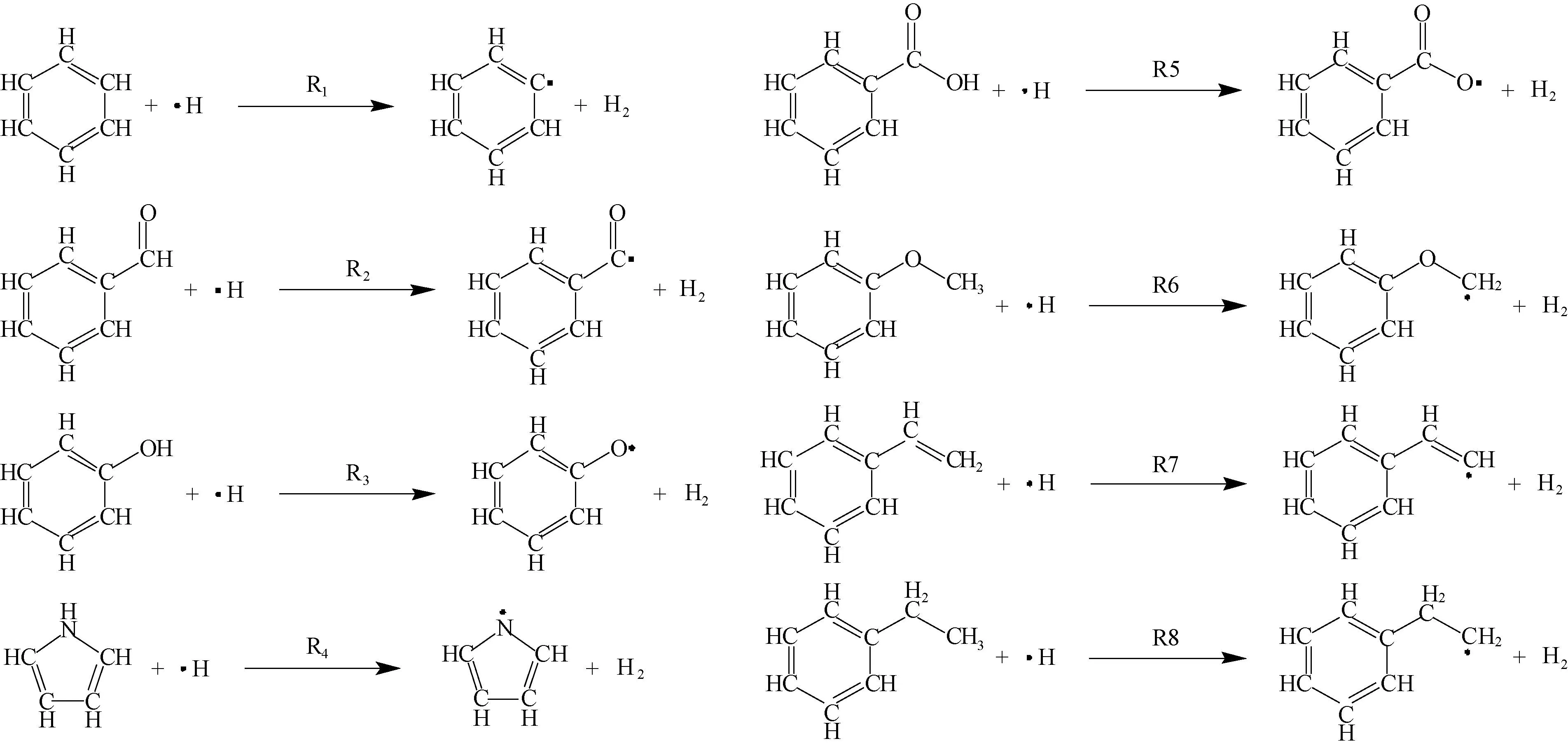
图3 不同模型H2生成反应图
由表4可得,苯和苯乙烯的H2生成反应常温下是吸热反应。除此之外,其他反应均为放热反应,放热量大小趋势为:苯甲酸<苯乙烷<四氢吡咯<苯甲醚<苯甲醛<苯酚。其中苯酚放热量最大,达到-91.265 kJ·mol-1. 反应自发性与焓变趋势一致,只有苯和苯乙烯反应呈现常温下不可自发发生,而其余模型ΔG<0,在常温下均能与H·发生反应。此外,结合反应平衡分析可知,上述模型中苯、苯乙烯和苯甲酸在常温下平衡常数K处于10-5~105,都无法完全反应。相较之下,其他模型K远远大于105,即在常温下便可完全反应,而其中又以苯甲醛和苯酚模型(R2与R3)数量级远远超过其余模型。以上从理论层面解释了H2的产生量与煤阶的关系[7].
显然,与前文分析一致,随着煤变质程度的降低,煤在自燃时更容易释放出H2. 另外,低变质程度的煤在H2生成反应中倾向于释放更多的热量,这又会加速煤自燃的进程。而平衡常数又指出低变质煤在与H·发生反应时会释放更多的H2.
有一点需要特别注意的是,H·并不是天然存在的,需要一定的激发条件(光、热、辐射等)才能产生,而煤体中热是最容易产生的激发条件。即当局部温度升高时,煤层中势必会产生大量的H·,进而释放远超正常情况下的H2,这一点可作为煤自燃程度的预测。
3 结 论
煤自燃过程中,H2的主要来源是H·与煤结构中的含氧官能团反应产生。此外,低变质程度煤倾向于在自燃时释放更多的H2和热量。因此,针对低变质程度煤层的自燃,可通过突变的H2浓度预测煤自燃程度。为更详细了解煤自燃机制,未来还应针对煤体中H·的产生机制展开研究。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
云南化工(2020年11期)2021-01-14
合成化学(2015年4期)2016-01-17
化工进展(2015年6期)2015-11-13
世界热带农业信息(2014年11期)2015-01-05
中成药(2014年11期)2014-02-28
河南科技(2014年11期)2014-02-27
化工生产与技术(2014年6期)2014-02-27
