
累代遗传的肝豆状核变性的临床与基因诊断分析
2024-01-31 10:59刘力生方明娟童广安王佳炜胡文彬
中风与神经疾病杂志 2024年1期
刘力生, 方明娟, 年 娜, 赵 雯, 童广安, 王佳炜, 胡文彬,
肝豆状核变性(hepatolenticular degeneration,HLD),又称Wilson 病,是由于ATP7B基因突变造成其编码的P 型铜转运ATP 酶缺陷引起肝铜跨膜转运障碍,使机体排铜障碍从而导致铜在肝、脑等多器官中过度蓄积,最终出现肝硬化、运动障碍、精神症状等[1]。HLD 是一种单基因常染色体隐性遗传性的、可治性的疾病,其发病率约1/30 000,人群携带率约为1/90,并具有区域差异,通常不会连续几代发病[2-4]。可能因为较高人群携带率,目前国外已有部分病例报道在同一家族内连续两代或多代中存在HLD,反映了“伪显性”遗传[5-8]。故而,2022 年美国肝病学会实践指导中明确提出对于HLD 确诊患者的一级亲属需进行ATP7B基因筛查[9]。目前国内尚无相关报道及研究,本研究针对我科近期收治的3 个累代遗传的HLD 家系进行了相关临床及基因的诊断,并进行总结分析,现报道如下。
1 资料与方法
1.1 一般资料
选择安徽中医药大学神经病学研究所附属医院神经内科二病区于2017 年6 月—2023 年7 月收治的HLD 患者3 例。收集调查其现病史、既往史、家族史等资料,并寻找其存在患病风险的父母、子女等一级亲属(见图1)。本研究经安徽中医药大学伦理委员会批准,取得每位参与者同意并签订知情同意书,采集家系成员的临床临检及生化指标、角膜K-F 环(kayser-fleischer ring,KFR)、腹部及头部影像学等临床资料,通过外周静脉血提取ATP7B基因检测。

图1 3个HLD家系遗传图谱
家系1:先证者(Ⅱ∶1),患者,男性,30岁,先后检查血小板减低、动作笨拙7 个月。2016 年11 月在当地医院检查发现“血小板、白细胞减低”,进一步检查腹部B超示:“肝硬化、脾肿大”,具体诊治不详。2017年2 月,家人发现其穿衣、持筷、系鞋带等精细动作笨拙,外院检查发现“血清铜蓝蛋白(ceruloplasmin,CP)减低”,予以“二巯基丙磺酸钠驱铜10 余天”,上述症状略有缓解。2017 年6 月5 日首次入住我科。家族史:否认父母近亲婚配,患者儿子(Ⅲ∶1)曾因“感冒”在当地医院检查“肝功能异常”。
家系2:先证者(Ⅲ∶2),患儿,女性,3 岁,检查发现转氨酶升高、CP 减低12 d。2021 年8 月6 日患儿在幼儿园入院体检时发现“肝功能:谷丙转氨酶(alanine aminotransferase,ALT)299 U/L,谷草转氨酶(aspartate aminotransferase,AST)184 U/L”,进一步检查“血清CP 0.1 g/L(参考范围0.16~0.45);肝炎病毒学指标及自身免疫性肝炎指标均正常”,遂于2021年8月17 日入我科。家族史:否认父母近亲婚配,患儿母亲(Ⅱ∶2)及小姨(Ⅱ∶4)有不明原因转氨酶升高情况,胞兄(Ⅲ∶1)体健,未行HLD相关筛查。
家系3:先证者(Ⅰ∶2),患者,女性,54 岁,渐进性头抖、四肢抖动16 年。2007 年起先后出现头部及四肢不自主抖动。至2013 年出现行走欠稳及言语含糊,就诊北京某医院考虑“HLD”,予以“驱铜”等治疗后上述症状明显改善。2023 年4 月初,因家庭矛盾出现情绪低落、夜间入睡困难,于2023 年4月23 日入我科。家族史:否认父母近亲婚配,患者儿子(Ⅱ∶1)曾检查血清铜蓝蛋白减低,近期体检有脂肪肝。
1.2 方法
1.2.1 一般检查项目 血清学指标(ALT、AST、血清CP)使用日立7180全自动生化分析仪检测,基础24 h尿铜排泄量(尿Cu)采用火焰原子吸收光谱法,仪器为北京瑞利分析仪器公司生产WFX-120B型原子吸收分光光度计。由我院专业人员在裂隙灯(YT2A型,苏州医疗器械厂)下进行KFR检查。使用Siemens ACUSON Sequoia超声(5C1探头)检查进行腹部彩超检查;对于家系1、3 先证者使用采用Philips Achieva 1.5T MRI扫描设备进行头部影像学检查。
1.2.2 基因检测 对先证者及其诊断存在疑问的一级亲属进行ATP7B基因二代测序(next generation sequencing,NGS),所有突变位点都通过多个数据库进行评估,包括HGMD 专业版、Clinvar、gnom AD 等数据库。使用Mutation Taster 和PolyPhen-2 软件检测分析所有可能致病突变的危害性。并对所有的突变位点进行ACMG(美国医学遗传学与基因组学学会)遗传变异分类标准和指南进行分类。最后,所有可能的致病性突变都用Sanger进行了测序,并通过家族谱系进行验证。诊断方法参照2021年《中国肝豆状核变性诊治指南》[10],并符合2001年莱比锡第8届国际HLD 会议提出的诊断标准(Leipzig计分系统)的积分 ≥ 4分[11]。
2 结 果
2.1 一般检查项目结果 家系1,Ⅱ∶1存在明显CP减低和尿Cu增高,ALT、AST正常,KFR阳性,腹部彩超提示肝硬化、脾大,头部MRI平扫提示脑干片状长T2信号,FLAIR呈稍高信号(见图2);Ⅲ∶1仅有轻度CP减低,但尿Cu增高,ALT水平轻度升高,KFR阴性,腹部彩超未见异常,遂进一步完善基因检测。
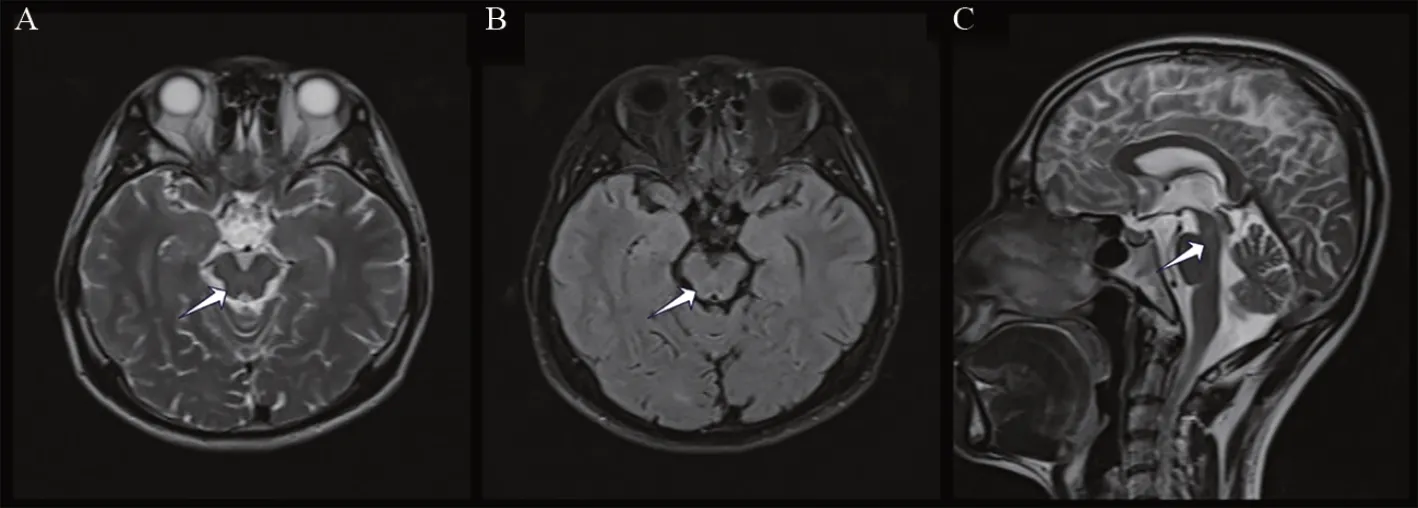
图2 家系1的Ⅱ∶1头部MRI平扫
家系2,Ⅲ∶2、Ⅲ∶1 均有CP 减低和尿Cu 增高,KFR 均阴性。Ⅲ∶2 的ALT、AST 水平明显升高,腹部彩超提示肝内钙化灶;但Ⅲ∶1 肝功能及腹部彩超均正常,遂进一步完善基因检测,结果显示与Ⅲ∶2基因型存在差异。因Ⅱ∶2、Ⅱ∶4 曾于外院检查CP 均为0.11 g/L,且均有不明原因转氨酶增高情况,而我院CP 仅轻度减低或正常,且Ⅱ∶4 尿Cu 正常范围,KFR均阴性,故同时进行基因检测。
家系3,Ⅰ∶2 存在明显CP 减低,尿Cu 不高,肝功能指标正常,KFR 阳性,腹部彩超提示肝硬化、脾稍大,头部MRI平扫提示脑干、双侧额顶叶皮质下、双侧放射冠区可见点片状长T2信号,FLAIR 呈高信号;脑室系统稍扩张,两侧大小脑半球脑沟裂增宽(见图3);Ⅱ∶1的CP明显减低,尿Cu高,腹部彩超提示脂肪肝,但肝功能正常,KFR 阴性,故予以完善基因检测。3个HLD家系一般检验资料(见表1)。
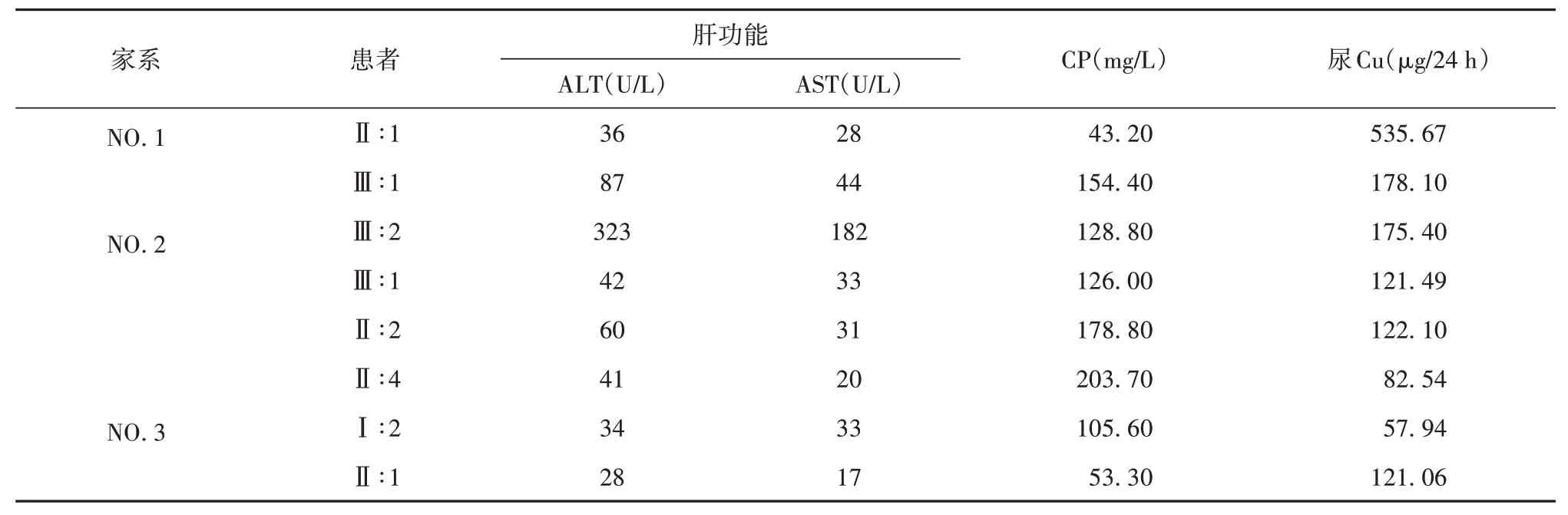
表1 3个HLD家系一般检验资料
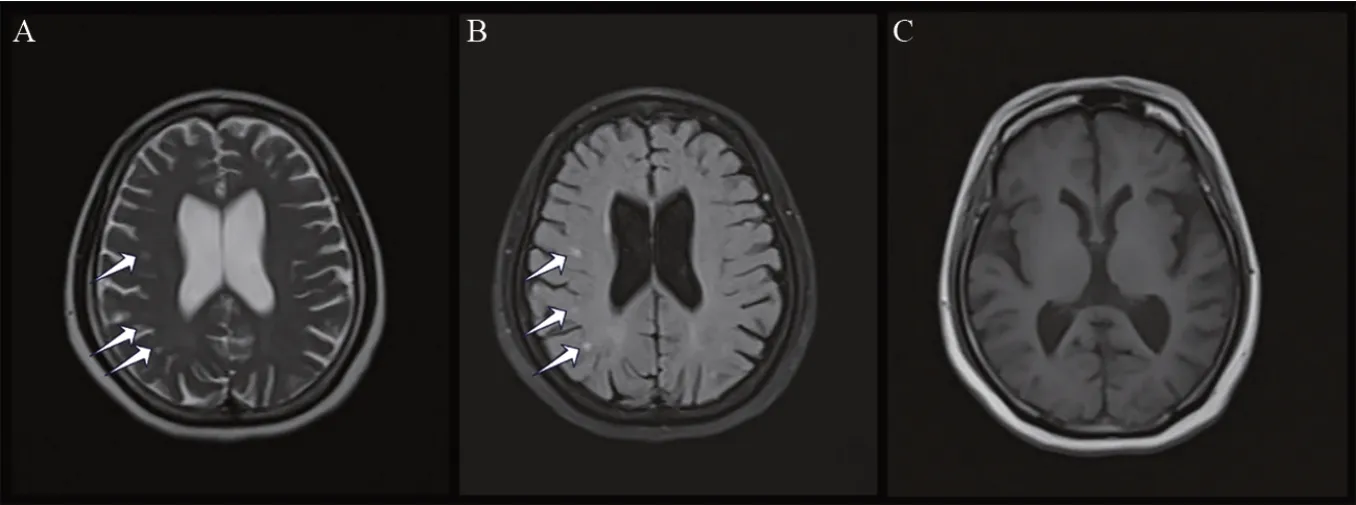
图3 家系3的Ⅰ∶2头部MRI平扫
2.2 基因检测结果 3 个家系共计发现六处杂合错义突变和一处缺失突变,分别是位于14 号外显子的c. 3121C > T p. Arg1041Trp、8 号外显子的c.2333G > T p.Arg778Leu、12号外显子的c.2975C > T p. Pro992Leu、13 号外显子的c. 2755C > G p. Arg919 Gly、11号外显子的c.2605G > A p.Gly869Arg和12号外显子的c.2790_2792delCAT p.Ile930del,均为已报道的HLD 常见致病位点。然而,位于2 号外显子的c. 482T > C p. Ile161Thr,目前在Clinvar 数据库有收录,致病性分级为Uncertain significance,HGMD 专业版暂未收录,ACMG 致病性分析为PP3 Moderate +PM2_support+PP1_suppor,分级亦为Uncertain significance;通过PolyPhen-2、Mutation Taster 软件分析该位点危害性结果均为:0.999(见表2)。

表2 3个HLD家系ATP7B基因突变位点
2.3 3 个HLD 家系诊断结果 根据Leipzig 计分系统:家系3 中Ⅱ:1 以“CP 53.3 mg/L”“尿Cu 121.06 μg/24 h”“ATP7B基因 c. 2333G>T p.Arg778Leu杂合突变”计算积分 ≥ 4分。最终,通过一般检查项目及ATP7B基因检测,家系1 中Ⅱ∶1、Ⅲ∶1,家系2 中Ⅲ∶2、Ⅲ∶1、Ⅱ∶2、Ⅱ∶4,家系3 中Ⅰ∶2、Ⅱ∶1 均确诊为HLD;家系2 中Ⅰ∶1、Ⅰ∶2、Ⅱ∶1、Ⅲ∶3,家系3 中Ⅰ∶1、Ⅲ∶1、Ⅲ∶2均被明确为HLD杂合突变携带者。
3 讨 论
HLD是一种由ATP7B基因突变引起的罕见的常染色体隐性遗传性铜代谢障碍性疾病[12]。1968 年,Sternlieb 和Scheinberg 首次估计了其患病率为5/100万[13]。随后的研究显示,德国每100 万人中有29 例HLD[14],日本每100万人中有33例HLD[15]。1984年,HLD 的全球患病率估计为3.3/100 000,经推算突变携带者频率为1/90[16]。国内最早由胡文彬对安徽省部分地区进行了两次流行病学调查,得出HLD 患病率为4.93/10 万~6.21/10 万[3,17]。而对封闭/孤立人群的研究显示:希腊卡利姆诺斯岛HLD 突变携带者频率达7/100,意大利撒丁岛的HLD 患病率高达1/2 707[18]。此外,在英国的一项分子研究预测携带两个致病ATP7B等位基因的个体的频率为1/7 026,突变携带者频率高达2.4/100[19];法国通过二代测序也发现HLD 人群杂合携带率达1/31[20]。因此,HLD 的患病率及普通人群突变携带率可能比以往认为的要高得多,当HLD 患者与杂合突变携带者婚配,这就意味着HLD 患者的一级亲属,如除兄弟姐妹外的父母、子代,患病的可能性不容忽视。
血清CP 的检测是目前临床诊断HLD 最常用的方法。根据最新Cochrane 数据库的系统评价结果显示,当以0.2 g/L 作为临界值时,CP 的敏感性为77.1%~99.0%,特异性为55.9%~82.8%;而当以0.1 g/L作为临界值时,总体的敏感性下降至65%~78.9%,但特异性提高至96.6%~100%[21]。然而,部分HLD患者的CP水平接近正常,本单位前期研究表明HLD患者CP 正常化与铜负荷过重以及肝损伤有关[22]。此外,在儿童HLD 的CP 水平研究中也发现,部分症状前期患儿CP水平是正常的[23]。本研究中,除家系1 的Ⅱ∶1 和家系3 的Ⅰ∶2 表现为典型的神经系统及肝硬化症状外,其余6例HLD 患者均为症状前期患者。其中1 例患者CP 在正常范围(16.7%),只有1 例患者CP水平低于0.1 g/L(仅占16.7%)。故而,不能单纯的以“CP 正常”便排除HLD 患者一级亲属的患病可能。
尿Cu 可以间接反映血清游离铜水平,有助于HLD 的诊断和治疗监测。当尿Cu>100 μg/24 h 时,对诊断HLD 的敏感性为50%~80%,特异性为75.6%~98.3%;对于无症状和儿童患者,当尿Cu>40 μg/24 h 时,对诊断HLD 的敏感性为78.9%,特异性为87.9%[21]。若能够排除其他慢性活动性肝病、肝硬化等疾病,尿Cu越高对HLD诊断价值越大。另外,尿Cu 水平会随驱铜治疗逐渐下降。本研究中,除家系3的Ⅰ∶2入院前曾进行驱铜导致尿Cu偏低外,其余患者尿Cu 均增高,符合诊断标准(100%)。因此,尿Cu测定对于筛查症状前期HLD具有较高的特异性。
根据研究显示,除胆汁淤积性肝病、部分胆道闭锁、自身免疫性肝炎等少见疾病外,KFR被广泛认为是HLD的一种特征性标志[24]。另有研究指出:在神经型HLD患者中KFR阳性率约72%~85.2%,而内脏型及症状前期患者中分别仅50%及20%~30%[25,26]。本研究结果显示,除家系1 和家系3 中的2 名先证者,亦为神经型患者的KFR 为阳性外,其余6名症状前期HLD 患者KFR 均为阴性,即阳性率为0%,因此KFR对于症状前期HLD患者的诊断敏感性较差。
目前已知的ATP7B基因致病位点达600 余种,其中以单核苷酸错义和无义突变最为常见,其次是插入/缺失,而剪接位点突变很少见[4]。在欧洲HLD患者人群中,最常见的突变为p.H1069Q[27],而我国人群的高频致病突变依次为p. R778L,p. P992L 和p.T935M[28,29]。随着人类基因组计划的完成和基因突变数据的快速积累,研究者可以有效地利用生物信息学软件来综合预测新突变的致病性[30]。其中,PolyPhen-2 是基于进化保守性与蛋白的三维结构,利用贝叶斯分类器计算后验概率来预测[31];Mutation Taster 是基于进化保守性、Grantham 矩阵评分、贝叶斯分类器来预测[32];二者在灵敏度、特异性、阳性预测值、准确度等多个指标参数与其他生物信息学软件对比,表现出较好的评估性能[30]。本研究中8例HLD患者均为复合杂合突变,共发现7个突变位点,其中p.R919G出现4次(25%),其次是p.R778L、p. G869R 出现3 次(18.8%),p. P992L、p. I930del 出现2 次(12.5%),p. R1041W、p. I161T 出现1 次(6.3%)。其中除p. I161T 外均为已报道HLD 致病位点。从家系3 中Ⅱ:1 发现的p. I161T 目前仅在Clinvar 数据库有收录,但致病性分级为“Uncertain significance”,HGMD 专业版未收录,ACMG 致病性分析为“PP3 Moderate+PM2_support+PP1_suppor”,分级亦为“Uncertain significance”;通过PolyPhen-2、Mutation Taster 软件分析该位点危害性分别为“PROBABLY DAMAGING”“disease causing”,评分均为:0.999。结合该患者已有的临床数据计算Leipzig评分≥4 分,故可诊断症状前期HLD,本研究进一步证实了p.I161T突变的致病性。
HLD 的临床异质性较大,患者发病年龄可从婴儿期至70 岁以上。患者可能出现各种类型的肝病、神经精神障碍等症状,亦可无任何临床症状[33,34]。对于典型的HLD 患者,根据最新的指南进行诊断并不困难[10,35],然而,对于部分临床表现不典型的患者,如症状前期患者,一方面,缺乏典型的肝、脑损害的症状、体征;另一方面,常规的临床指标如CP、KFR 等也可能与典型HLD 患者表现不同;这些均增加了诊断难度。有研究表明,血清转氨酶升高是儿童或症状前期HLD 诊断的重要线索[23]。本研究中,6 例症状前期患者有5 例出现不同的程度转氨酶升高情况(83.3%)。随着HLD 基因的克隆以及基因诊断技术的应用和进步,在判断临床表现和实验室检查结果不典型的HLD 疑诊患者以及有HLD 先证者的家系成员是否患病方面,基因诊断有着十分重要的价值[36]。
HLD 是一种单基因常染色体隐性遗传疾病,携带者没有任何症状,通常被认为不会发生累代遗传,然而,由于HLD 致病基因ATP7B基因突变在人群中的高携带频率,我们进行了上述研究,以展示HLD的“伪显性”遗传特征,并明确了p.Ile161Thr 突变的致病性。HLD 是一种为数不多的可治疗的遗传病,早期诊断对于确保患者能够早期接受充分的治疗、避免进展为神经型和肝硬化至关重要。临床医师可通过肝功能、CP、尿Cu 等临床指标初步评估HLD 患者父母及子女的HLD 患病风险,必要时进行基因检测明确诊断。同时,建议HLD 患者生育前对配偶进行基因检测,以避免累代遗传的发生。然而,本研究因病例数有限,研究结果可能有失偏颇,待后期继续收集案例进一步分析、验证。
伦理学声明:本研究经安徽中医药大学神经病学研究所附属医院伦理委员会批准[批号:2023伦字(27)号],患者均签署知情同意书。
利益冲突声明:所有作者均声明不存在利益冲突。
作者贡献声明:刘力生负责论文设计、起草论文;方明娟、赵雯负责收集临床资料、绘制图表;年娜负责基因分析;童广安、王佳炜负责文献收集、拟定写作思路;胡文彬负责论文修改、指导撰写文章并最后定稿。
猜你喜欢
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
生物学通报(2019年3期)2019-02-17
湖南畜牧兽医(2016年3期)2016-06-05
兽医导刊(2016年12期)2016-05-17
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
郑州大学学报(医学版)(2015年2期)2015-02-27
当代畜禽养殖业(2014年7期)2014-02-27
