
环辛四烯的结构、芳香性及其异构化反应
2024-03-04 09:01凡维钱旭清王宇轩杨佳璐张孙秋骏王信凝韩俊骁鹿成龙张冬菊
大学化学 2024年1期
凡维,钱旭清,王宇轩,杨佳璐,张孙秋骏,王信凝,韩俊骁,鹿成龙,张冬菊
山东大学化学与化工学院,济南 250100
环辛四烯(Cyclooctatetraene,COT),又名1,3,5,7-环辛四烯,无色至淡黄色液体,具有粘膜刺激性,有一定毒性,不溶于水,溶于乙醇、乙醚、丙酮、苯等有机溶剂,熔点−4.7 °C,沸点142–143 °C。环辛四烯属于环状多烯类化合物,在大学有机化学[1,2]和结构化学[3,4]课程中均有介绍,文献中也有相关讨论[5,6]。教学中我们发现,学生对环辛四烯结构和性能的理解不够深入,存在一些模糊不清甚至错误的认识。学生经常提出的问题包括:为什么环辛四烯的稳定结构不是平面构型而呈盆型结构,而环辛四烯的阴离子却呈现平面构型,该分子的基态是单重态还是三重态,环辛四烯是非芳香性体系还是反芳香性体系,环辛四烯如何进行异构化反应等。对这些问题回答,涉及多个知识点的综合应用,包括处理π电子体系的Hückel分子轨道(Hückel Molecular Orbital,HMO)法、π电子体系芳香性的Hückel规则[7]和Baird规则[8]、离域能、Jahn-Teller畸变效应[9]等重要基础理论和化学概念。为加深学生对这些化学基础知识和基本理论的理解,围绕环辛四烯的结构、芳香性、异构化反应等问题,我们凝练了一系列科学问题,设计了该教学案例,并以开放课题形式鼓励本科生参与分析问题、解决问题,引导学生深刻理解物质结构与性能的内在联系,强调借助量子化学计算结果加深对化学基础知识和基本理论的理解和应用。
1 实验结果
环辛四烯首先由德国化学家Willstätter等[10]在1905年通过化学合成得到(图1a),后来Stinson等[11]发现存在于天然产物中,并在一些真菌中分离出来。图1a给出了Willstätter等[10]以含有八元环骨架的伪石榴碱为原料制备环辛四烯的合成路线:伪石榴碱(A)在乙醇中被钠还原为醇,继而在酸性条件下脱水,形成八元环骨架的第一个C=C双键(结构B);胺的甲基化和季铵盐的Hoffmann消除反应,依次构建第二和第三个C=C双键(结构C和D);溴对多烯的1,6-共轭加成和二甲胺的亲核取代,形成双取代双胺中间体(E);最后再次经历胺甲基化和Hoffmann消除反应,得到环辛四烯。该方法由于过程复杂、产率极低(0.75%)等原因而未得到重视,甚至由于难以重复而被质疑。直到1940年德国化学家Reppe等[12]发现了乙炔四聚法的合成路线(图1b),才逐渐实现了环辛四烯的规模化生产。如图1b所示,Reppe使用二价镍盐催化剂,在20 atm (2026.5 kPa)下,将乙炔通入碳化钙在四氢呋喃中反应,实现了乙炔的四聚反应,得到了高产率(90%)的环辛四烯。由于操作简单、产率高,乙炔四聚法自发现以来一直是合成环辛四烯的最主要方法。

图1 以伪石榴碱(a)和乙炔(b)为原料合成环辛四烯
环辛四烯具有典型的不饱和烃的性质,稳定性不高,容易发生氧化、聚合、加成等反应,主要作为合成其他化合物(如醇、酸和酯等)的原料和中间体,在化学、食品、农药、医药等工业均有应用。环辛四烯容易被氧化生成爆炸性的有机过氧化物,市售的环辛四烯通常加入少量的氢醌作稳定剂。使用环辛四烯之前应当检验过氧化物的存在并妥善处理,避免事故发生。
环辛四烯的二价阴离子(cyclooctatetraenyl dianion,COT2−)由环辛四烯衍生得到。1960年Katz[13]首先证实了环辛四烯二价阴离子的存在,他们通过环辛四烯与金属钾在液氨中的反应,获得了K2COT。与中性分子不同,COT2−具有较高的稳定性,不易发生通常烯烃的氧化、聚合和加成反应。
2 几何结构
文献中报道的环辛四烯构型的畸变构型主要有三种,如图2中结构I、II、III所示,根据四个双键的顺反构型分别命名为(Z,Z,Z,Z)型(盆型)、(Z,E,Z,Z)型(扭曲型)、(Z,E,Z,E)型(椅型),三个结构分别具有D2d、C2、C2h对称性[14]。1950年前后,Kaufman等[15]通过X光衍射实验证实了环辛四烯中的C―C键有两种不同的键长;后来Leutwyler课题组[16]使用时间分辨飞秒相干光谱证实了盆型结构是环辛四烯最稳定的构型,测定了分子中两种不同的C―C键长(0.147和0.134 nm),分别接近C―C单键和双键的键长。环辛四烯二价阴离子则为平面型分子,具有D8h对称性,分子中C―C键长均为0.141 nm[17]。
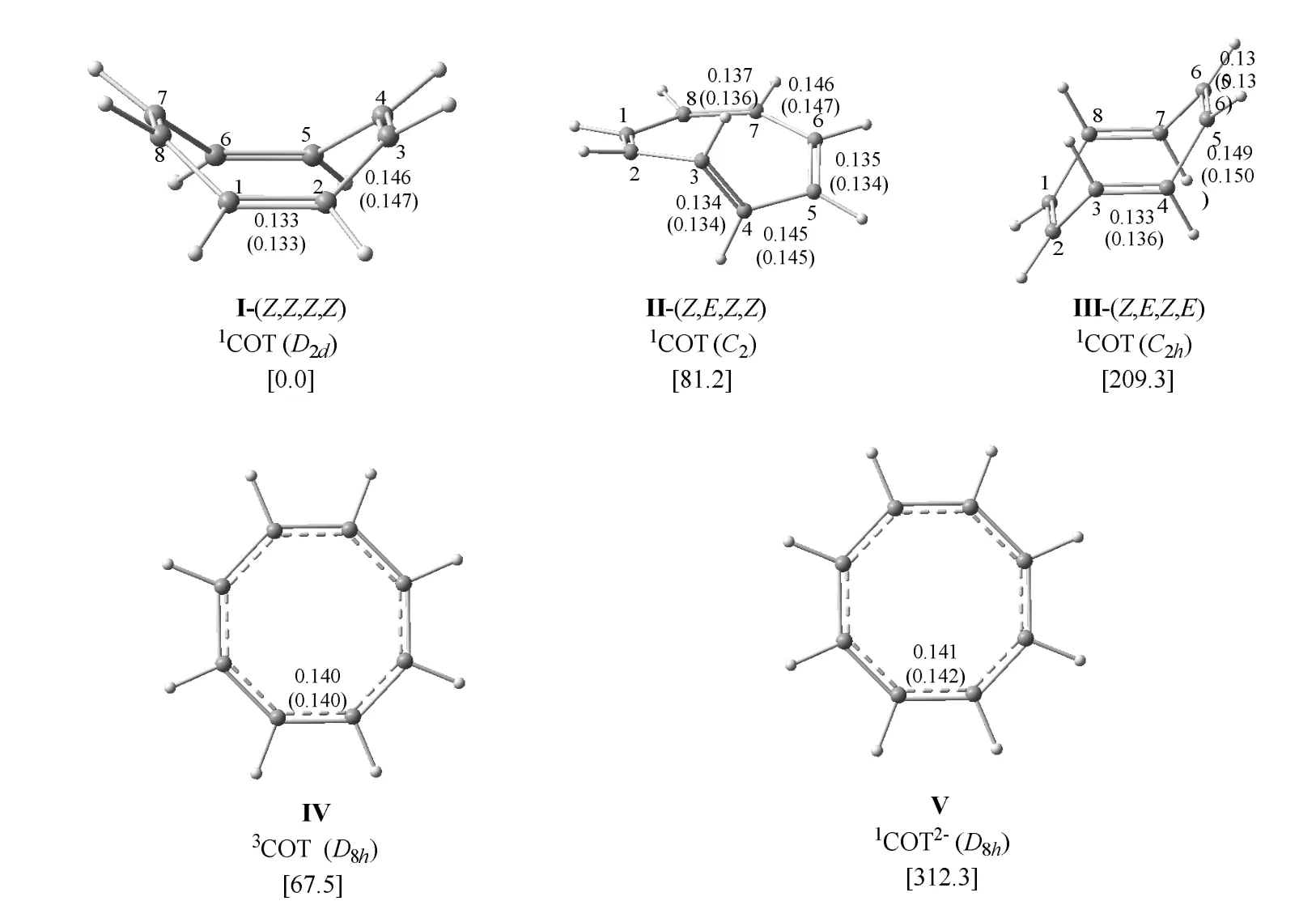
图2 DFT方法计算的环辛四烯单重态(I, II, III)、三重态(IV)及其二价阴离子(V)的几何构型(键长:nm,圆括号中的值为实验结果[18,19])和相对能量(方括号中的值,kJ·mol−1)
为加深对环辛四烯及其阴离子结构的理解,我们对图2中几个结构进行了量子化学计算,使用GaussView 6.0[20]构建图2中的四个结构模型,在M06[21]/6-311G(d,p)[22]水平上进行了构型优化和振动分析,计算中未施加对称性限制,全部计算使用Gaussian 09程序[23]完成。优化的构型参数在图2中列出。结果表明,四个结构的对称性与实验或以前的理论结果完全一致,结构参数也与文献符合较好(图2)。环辛四烯三个异构体的相对能量也在图2中列出,盆式构型(D2d)最稳定,是环辛四烯的基态结构。扭曲型(C2)和椅型异构体(C2h)的相对能量分别为81.2和209.3 kJ·mol−1,两个结构的不稳定性可从计算的结构参数得到合理解释。如图2所示,对于盆型结构I,每个双键均与其两端的碳原子共平面;扭曲型结构II中,对于反式构型的双键,C2―C3―C4―C5二面角畸变到130°;椅型结构中,二面角C2―C3―C4―C5和C6―C7―C8―C1分别畸变到121.1°和−121.1°,这些较大的构型畸变导致体系能量明显升高。对于环辛四烯的二价阴离子(图2,结构IV),构型优化给出平面正八边形结构,呈D8h对称性,C―C键长均为0.143 nm,也与实验值符合较好。振动分析表明,这些结构均无振动虚频,表明它们是势能面的稳定点。
3 电子结构
上述实验和计算的结构参数表明,环辛四烯中虽然类似共轭多烯,C―C单、双键交替分布,但没有π电子的离域,而其二价阴离子则呈现典型的共轭体系特征,C―C键长完全平均化,键长介于单、双键之间。为理解这些几何结构特征,需要弄清其π电子结构。Hückel分子轨道法(HMO)是近似求解共轭π电子薛定谔方程的近似方法,在结构化学教材[3,4]中有详细介绍。环辛四烯及其二价阴离子的碳环骨架相同,根据HMO方法,类似于教材中对丁二烯和苯分子的处理,将π电子波函数写为8个碳原子p轨道的线性组合,通过线性变分法求解久期方程,分别得到π电子能级和波函数,具体求解过程参考结构化学教材[3,4],这里不再赘述,仅给出求解的8个π分子轨道能级。如图3所示,ψ1,ψ2,ψ3为成键轨道,ψ4和ψ5为一对简并的非键轨道,ψ6,ψ7,ψ8为非键轨道。八个π分子轨道的能级用α和β表示,即E=α+xβ,其中,φi和φj为碳原子pz轨道,i、j为碳原子编号,α为库仑积分,近似等于碳原子2pz轨道的能量,而β为交换积分,其值为一小于零的负值(−2.4 eV)。x通过求解久期方程得到,其值越负,轨道能级越低。
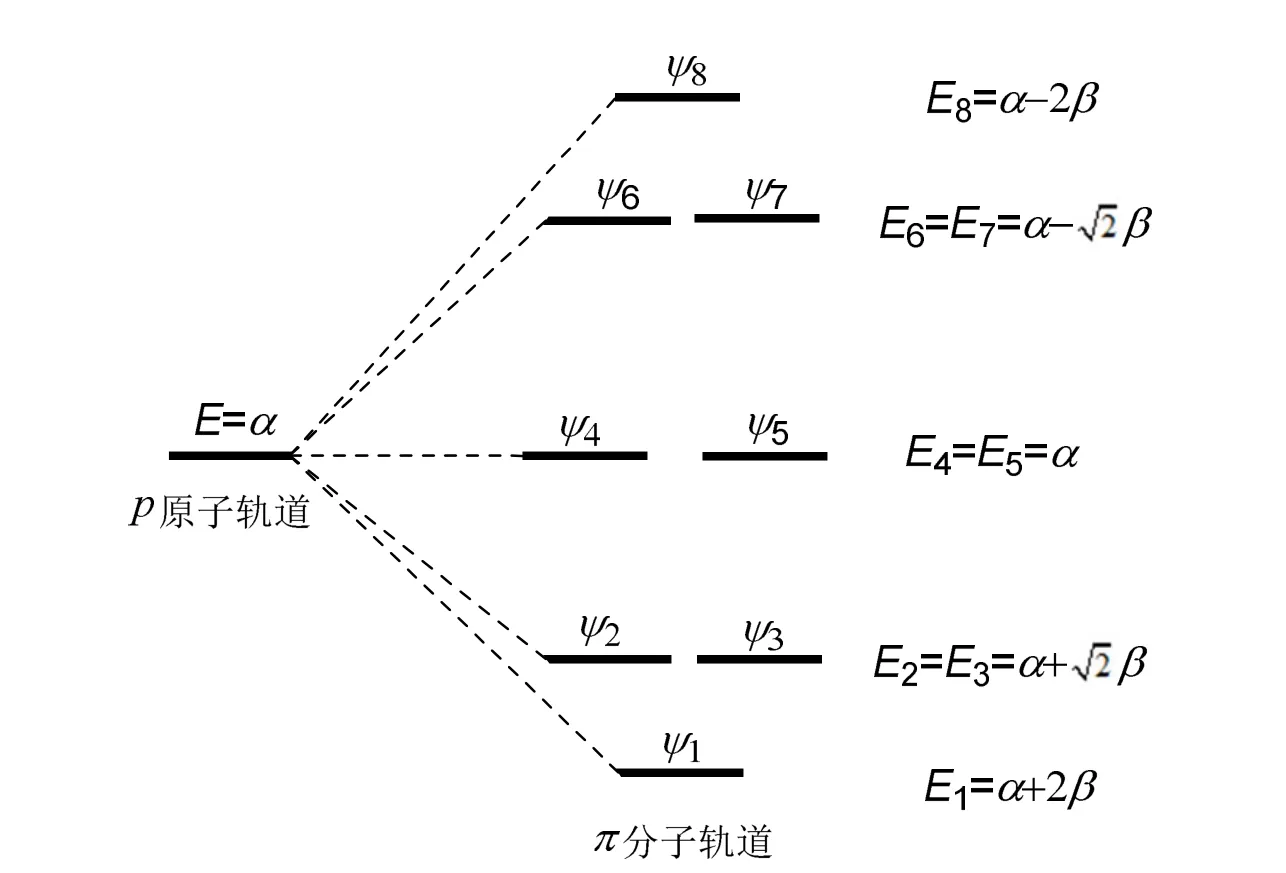
图3 Hückel分子轨道法处理假想的“平面八元轮烯”得到的π分子轨道能级示意图
对于环辛四烯,假定分子为Hückel型八元环平面分子,8个π电子在图3所示的分子轨道中布居时,将会出现两个π电子分占两个简并的非键轨道(ψ4和ψ5),并保持自旋平行,这样得到的是体系的三重态。Jahn-Teller畸变效应指出,对于非线性分子,电子分占简并能级的布居状态是不稳定的,将通过构型畸变降低分子对称性消除简并,到达稳定状态。因此,环辛四烯的三重态(平面构型)不是最稳定的结构,将通过构型畸变消除简并,到达稳定的单重态。文献中报道的环辛四烯构型的畸变构型主要有三种,如图2所示,其中盆型构型是分子的基态构型,这与其电子数不满足4n+ 2 (n为整数)芳香性规则、体系不具有Hückel芳香性结果一致。这表明用Hückel分子轨道法处理环辛四烯,并不能得到合理的结果。对环丁二烯和其它4n个π电子体系的处理也会得到类似的结果,均会得到一对简并的非键轨道,导致π电子体系的三重态布居,与实验测定的单重态基态不符,这是Hückel分子轨道法的局限性[24]。
环辛四烯的二价阴离子有10个π电子,两个非键轨道被全充满,是闭壳层结构,因此是分子的稳定状态,并且其π电子数满足Hückel芳香性规则,体系呈现平面结构。
4 芳香性、反芳香性、非芳香性的讨论
关于环辛四烯的芳香性(aromaticity)问题,文献中有不同描述,有的将其描述为“反芳香性”(antiaromaticity)体系,有的则描述为“非芳香性”(nonaromaticity)体系,概念较为混乱。首先需要澄清“芳香性”“反芳香性”和“非芳香性”等基本概念。“芳香性”与“反芳香性”是一对描述“平面、环状、共轭”体系稳定性的术语,二者的唯一区别是体系中π电子数目不同。芳香性体系,π电子数为4n+ 2,符合Hückel规则,具有特殊的稳定性,如苯、吡啶、萘等分子;而反芳香性体系,其π电子数为4n,体系不稳定,如环丁二烯、环戊二烯阳离子、并环戊二烯等体系。“非芳香性”意指体系不同时满足“平面、环状、共轭”等三个条件,不适宜用芳香性、反芳香性的概念描述体系的稳定性,这样的体系是“非芳香性”的。可以看出,除非特别指明(如周环反应中涉及的Möbius芳香性),“芳香性”“反芳香性”和“非芳香性”中的“芳香性”通常均指Hückel芳香性。
如上所述,环辛四烯的基态(单重态)为盆式构型,不是平面型分子,不同时满足“平面、环状、共轭”条件,并且从优化的构型可以看出,分子中两类不同的C―C键长分别为典型的共价单键和双键,是典型的非共轭体系,因此环辛四烯是非芳香性体系,虽然其π电子数符合4n规则,但不能将其描述为“反芳香性”体系。
应该注意,上述关于“平面、环状、共轭”型分子芳香性的4n+ 2规则和反芳香性的4n规则,仅适用于分子基态(单重态)的情况。对于“平面、环状、共轭”型分子第一激发态(三重态)的情况,体系的芳香性和反芳香性规则恰好相反,即π电子数为4n+ 2的体系,具有反芳香性;而π电子数为4n的体系则具有芳香性。与基态的Hückel规则对应,确定“平面、环状、共轭”型环烯体系三重态芳香性的规则被称为Baird规则[25,26],由Baird[8]在1972年提出。Baird规则指出,闭合环状三重态平面型的共轭多烯,当其π电子数为4n时,具有芳香性。环辛四烯的三重态满足Baird规则,具有Baird芳香性,为平面型分子。为证实这推测,我们也在M06/6-311G(d,p)水平对环辛四烯的三重态进行了量子化学计算,结果发现,从盆型、扭曲型或椅型任一结构开始,环辛四烯的三重态总是收敛到平面正八边型构型,与其二价阴离子类似,具有D8h对称性,所有C―C键长均为0.140 nm。与单重态盆型结构相比,环辛四烯三重态的相对能量为67.51 kJ·mol−1。实际上环辛四烯的三重态,正是上述HMO法求解环辛四烯π电子薛定谔方程的结果,处理时将体系视为正八元环结构,得到的是体系的三重态。
三重态环辛四烯的芳香性也可通过计算离域能得到验证:
离域能为负值,表明三重态环辛四烯π电子能量低于4个孤立C=C双键的π电子能量,具有芳香性。
为了进一步证实三重态环辛四烯以及环辛四烯二价负离子的芳香性,我们通过量子化学计算给出了评估体系芳香性的磁学判据,即核独立化学位移(Nucleus-independent chemical shifts,NICS)。该判据由Schleyer等[27]在1996年提出,它采用环体系中心的化学位移作为环电流探针,计算体系环电流对探针中心的屏蔽作用。所以,体系的NICS值成为其芳香性和反芳香性的指标之一,其值越负,表明体系对磁场屏蔽越强,体系的芳香性越强。本文选用NICS(1)zz指标比较体系芳香的相对强弱[28],即体系平面上方1 Å (1 Å = 0.1 nm)处垂直于环平面方向的屏蔽张量分量值的负值。在M06/6-311G(d,p)水平,计算的环辛四烯基态三重态、环辛四烯二价阴离子的NICS(1)zz值均为负值(使用Gaussian程序默认的GIAO方法[29]进行计算),分别为–33.19和–40.46 ppm。作为参照,我们也对苯分子(典型的芳香性体系)进行了计算,其NICS(1)zz值为–30.22 ppm。显然,环辛四烯三重态与环辛四烯二价阴离子均呈现显著的芳香性。
5 异构化反应
有机化学教材[1]中提到,在70 °C和300 nm的光源照射下,环辛四烯气体几乎可以定量地异构化为半瞬烯(semibullvalene) (图4a)。实际上,环辛四烯不仅在光照条件下可以发生异构化反应,而且也可在基态势能面上发生热异构化反应[30]。如图4b所示,100 °C时,环辛四烯异构化为双环[4.2.0]辛-2,4,7-三烯(VI),而在300 °C时,则可异构化为半瞬烯(VIIa/VIIb)。这里我们以环辛四烯的热异构化反应为例,研究其反应机理,解释为什么不同温度下得到不同的异构化产物。计算结果示于图5。

图4 环辛四烯在光照(a)和加热(b)条件下的异构化反应
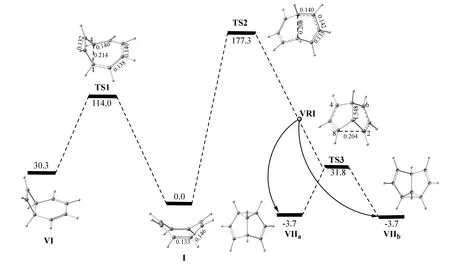
图5 环辛四烯典型热异构化反应的势能剖面图(相对吉布斯自由能:kJ∙mol−1)
双环结构VI的形成是一个基元过程,该反应类似于1,3-二烯烃的电环化反应,在加热条件下,轨道对称性允许C1和C4发生顺旋关环,形成环丁烯骨架,分子的其余部分仅发生扭曲,不涉及键的断裂和形成。反应经历的过渡态为TS1,根据其结构特征,可以将其描述为一个四元环过渡态,其中,C1-C4 (0.214 nm)形成σ键,同时C1-C2和C3-C4由双键形变为单键,C2-C3由单键变为双键。该异构化过程需要克服的能垒为114.0 kJ·mol−1,与文献报道结果[31](117.6 kJ·mol−1)符合较好。对于半瞬烯(结构VIIa/VIIb)的形成,计算表明,反应经历TS2和TS3两个过渡态,其中TS2具有C2对称性,对应C1-C5键的形成,该过程需要克服的能垒为177.3 kJ∙mol−1,与文献值[32](176.6 ± 0.84 kJ·mol−1)基本一致。应该注意的是,内禀反应坐标(IRC)计算证明,沿产物方向TS2连接的是过渡态TS3,而不是通常的势能面上的局域最小点。TS3呈C2v对称性,其虚频对应的振动模式表明该结构是一个Cope重排过渡态,连接两个半瞬烯VIIa和VIIb,其中C4-C6/C2-C8距离为0.204 nm。这是分叉势能面[33,34]的特征,在TS3之前,势能面上存在一个“谷脊拐点(valley-ridge inflection point,VRI点)”,在该点附近反应路径发生分叉,分别得到VIIa和VIIb。本文计算的半瞬烯Cope重排的能垒为35.5 kJ·mol−1,表明两个半瞬烯异构体容易相互转化。
从图5可以看出,TS1是决定反应速率的过渡态,从环辛四烯到半瞬烯的异构化反应,需要克服的总能垒(177.3 kJ·mol−1)远高于异构化为双环结构I的能垒(114.0 kJ·mol−1),仅在高温下才能发生。这些计算结果与实验观测相符,即温度相对较低时(100 °C)异构化为双环结构VIIa,而在较高温度下(300 °C)则异构化为半瞬烯VIIb。
6 结语
本文以物质结构–性能关系为主线,通过Hückel分子轨道法和量子化学计算系统讨论了环辛四烯的几何构型、电子结构、芳香性、异构化反应等内容,明确了其基态为单重态、呈盆型结构、具有D2d对称性,是非芳香性分子;分析了Hückel分子轨道法处理4n型π电子体系基态结构的局限性;阐明了环辛四烯二价阴离子的Hückel芳香性以及环辛四烯三重态的Baird芳香性,并基于计算的独立核化学位移指标,明确了其芳香性的内禀属性;给出了环辛四烯典型热异构化反应的势能剖面,合理解释了实验观测结果:高温下得到半瞬烯产物,而相对较低的温度下得到双环产物。本文结果为加深对环辛四烯结构和性能的理解提供了重要的理论支持,可用作教学参考资料,助力学生进阶学习。
猜你喜欢
大学化学(2021年8期)2021-09-26
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
山东化工(2020年5期)2020-04-07
石油石化绿色低碳(2019年6期)2019-01-14
北京航空航天大学学报(2017年10期)2017-04-20
化工进展(2015年3期)2015-11-11
华东理工大学学报(自然科学版)(2015年3期)2015-11-07
厦门大学学报(自然科学版)(2014年2期)2014-08-06
航天返回与遥感(2014年4期)2014-07-31
无机化学学报(2014年4期)2014-02-28
